











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
Se define ictiosis como un conjunto de alteraciones de la cornificación de la piel, de carácter monogénico (mendeliano), determinadas genéticamente, que se caracterizan por presentar hiperqueratosis o descamación, o ambas, y que en su fase inicial asocia ampollas. En la etapa neonatal únicamente son de carácter hereditario1,2.
La clasificación de las ictiosis hereditarias se basa en las características clínicas, considerando aspectos fisiopatológicos y moleculares, pudiéndose dividir según exista o no compromiso extracutáneo en sindrómicas y no sindrómicas, respectivamente (Tabla 1 y Tabla 2).
La ictiosis epidermolítica es una patología poco frecuente en la infancia, se estima en 1/300.000 nacimientos, siendo ésta la forma más prevalente dentro del grupo de las queratinopaticas3-5. En Uruguay, no existen datos publicados que permitan conocer la incidencia de esta enfermedad.
Presenta una herencia autosómica dominante, con penetrancia variable y se origina por mutaciones en los genes KRT1 y KRT10 que codifican las citoquinas 1 y 10 respectivamente3,6.
Las manifestaciones clínicas son muy variables. Al nacimiento, los pacientes suelen presentarse eritrodérmicos con formación de ampollas, erosiones y descamación hasta el mes de vida, por lo que es difícil de diferenciar de la epidermolisis bullosa. Luego comienza la hiperqueratosis a hacerse visible en codos y rodillas, volviéndose más evidente a los tres meses. Posteriormente, escamas verruciformes de color marrón grisáceo cubren la mayor parte de la piel, pliegues y zonas intertriginosas. Palmas y plantas pueden presentar distintos grados de afectación, siendo más grave en pacientes que tienen mutación KRT1. Un olor corporal desagradable se asocia frecuentemente con formas severas de este trastorno debido a la magnitud del espesor cutáneo, maceración y crecimiento excesivo de bacterias2,7.
El diagnóstico se realiza por las características clínicas, histológicas y alteraciones genéticas6. En la histopatología se observa una marcada hiperqueratosis con apoptosis de queratinocitos suprabasales (hiperqueratosis epidermolítica), que conduce a la formación de ampollas8.
El manejo de estos pacientes debe ser multidisciplinario, involucrando a neonatólogos, genetistas, dermatólogos y pediatras para lograr un manejo integral en cuanto a cuidado de la piel, curación y prevención de heridas, disminuyendo los riesgos de tener una barrera cutánea alterada y lograr un mejor pronóstico en estos pacientes.
El objetivo de esta presentación es comunicar el caso clínico de un neonato con una forma rara de ictiosis, como es la epidermolítica, con antecedentes familiares de primer grado de esta enfermedad y diagnóstico prenatal, describir su evolución posnatal y destacar la importancia del manejo por un equipo multidisciplinario.
Caso clínico
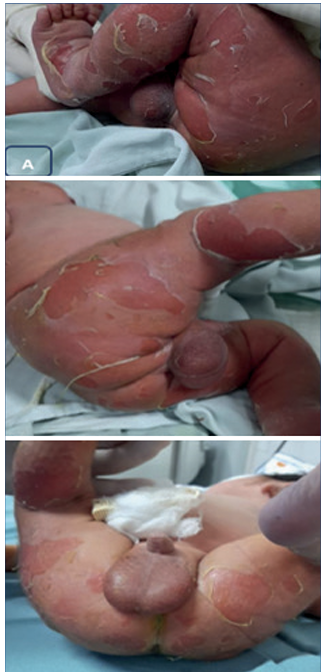
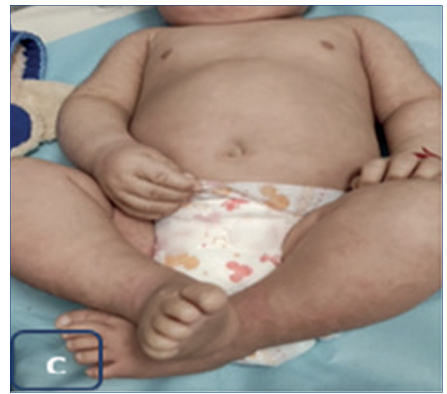
Neonato de sexo masculino, de término, macrosómico, grande para la edad gestacional, vigoroso, nacido en el interior del país donde se recepcionó como recién nacido sano. Se constató, mientras se seca y estimula, decolamiento extenso cutáneo (signo de Nikolsky positivo). Es trasladado a los cuatro días de vida a la unidad de cuidados intensivos del Centro Hospitalario Pereira Rossell. Se trata de un neonato con antecedentes familiares del padre con diagnóstico de ictiosis epidermolítica y diagnóstico prenatal por estudio genético que confirmó la mutación en el feto (C.467G Ap (Arg 156 His) asociada al gen KRT10). En sala de cuidados intensivos de neonatología, el examen físico mostró una dermatosis diseminada erosiva en áreas de fricción (Figura 1). Lesiones erosivas y decoladas en dorso alto interescapular, en miembros superiores a nivel de codos y en miembros inferiores en glúteos, región inguinal y cara interna de muslos. Presentaba una única ampolla de 0,5 mm de diámetro con contenido claro localizada en abdomen. No presentó áreas de exudación ni de sobreinfección, como tampoco compromiso mucoso ni faneral.
Fue ingresado en el sector de cuidados intermedios. Se realizó una manipulación manual mínima. Se colocó sobre un colchón de sábanas estériles. La higiene se realizó con limpiador tipo Syndet, secando con toques por oposición. Se utilizaron pañales con elástico recortados con vaselina en los bordes. Se cubrió la piel con apósitos de tul impregnados con excipiente glicólico hidrosoluble o gasas de vaselina estéril. Recibió analgesia vía oral durante las curaciones. Se alimentó con pecho directo exclusivo9,10. A las 48 horas de la valoración inicial, presentó a nivel interdigital en mano derecha, tórax y muslo izquierdo, lesiones pustulosas con halo eritematoso, se extrae hemocultivo y exudado cutáneo y se inició antibioticoterapia intravenosa con vancomicina, que se suspende a las 48 horas con hemocultivo sin desarrollo. Exudado cutáneo desarrolla Staphylococcus aureus, recibe tratamiento tópico con buena evolución (Figura 2).
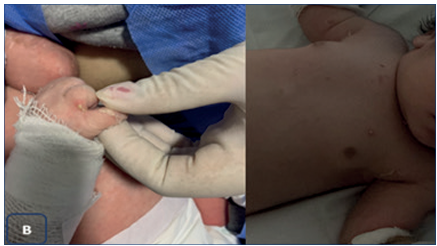
Figura 2 Nivel interdigital en mano derecha, tórax y muslo izquierdo, lesiones pustulosas con halo eritematoso (producidas por Staphylococcus aureus).
Se realizó biopsia cutánea bajo estrictas condiciones de asepsia y se envió muestra para estudio histopatológico con HyE, donde se observa hiperqueratosis ortoqueratósica con queratinocitos apoptóticos por encima de la capa basal (hiperqueratosis epidermolítica), que conduce a la formación de ampollas, compatible con ictiosis epidermolítica (Figura 4).
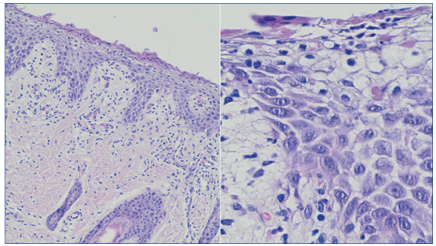
Figura 4 Histopatología con HyE: leve hiperqueratosis, epidermis acantopapilomatosa con queratina irregular eosinófila, vacuolización de la capa granulosa, gránulos de queratina no conservados y gránulos eosinófilos irregulares en sector superior (hiperqueratosis epidemolítica). Espongiosos. Dermis papilar, edema e infiltrado inflamatorio mixto con leucocitos, neutrófilos y eosinófilos.
El paciente fue dado de alta a los 20 días de vida, continuando en seguimiento en policlínica de dermatología y en el Centro de Referencia Nacional de Defectos Congénitos y Enfermedades Raras (CRENADECER).
Discusión
El caso relatado involucra una patología poco usual, una enfermedad de las catalogadas “raras” por su baja o variable prevalencia en la población (1/200.000-300.000), lo que la hace poco sospechada en la práctica clínica, pero de gran importancia dada su elevada morbimortalidad en la etapa neonatal. En Uruguay, no hay estudios que permitan conocer la incidencia de esta enfermedad.
En el caso presentado se confirma la enfermedad por estudio genético en el feto, presentando la misma mutación que su padre: C.467G Ap (Arg 156 His) asociada al gen KRT10, por presentar una herencia autosómica dominante con penetración completa; sin embargo, sabemos que existe una alta tasa de mutaciones esporádicas, en hasta un 50% de los casos no se encuentran antecedentes familiares. Numerosos genes están implicados en la cornificación, siendo su correcto funcionamiento crucial para mantener una barrera cutánea funcional. Este tipo de ictiosis se produce por mutación en los genes que codifican las queratinas epidérmicas suprabasales, KRT1 y KRT10. En el caso de nuestro paciente la mutación se encuentra en este último gen. Las queratinas defectuosas llevan al mal ensamblaje de los queratinocitos con la consiguiente formación de ampollas y, por otro lado, el defecto en la barrera cutánea estimula la producción de queratinocitos, resultando en una hiperqueratosis por aumento de recambio1,5,8.
El neonato presenta al nacimiento fragilidad cutánea, ya que al realizar maniobras de secado y estimulación presenta áreas extensas de decolamiento dejando expuestas áreas eritematosas. Esto se debe a que las queratinas defectuosas forman filamentos intermedios alterados que conducen a la lisis del citoesqueleto celular produciendo la formación de ampollas que al secar se rompen y generan dichas lesiones.
El diagnóstico se basa en las características clínicas, histológicas y genéticas. Se han logrado avances significativos en los últimos años gracias a la tecnología basada en la secuenciación de ADN y se siguen estudiando nuevas formas de estudio genético. De estar presente el estudio genético, permite un diagnóstico más certero y poder brindar opciones terapéuticas y pronóstico, así como consejería genética familiar, no siempre disponible en las instituciones1,2,6. La importancia de contar con el diagnóstico prenatal, como ocurre con nuestro paciente, es que se puede recepcionar y manejar por un equipo multidisciplinario, aplicando los cuidados para piel frágil. Haciendo referencia al caso clínico, al tratarse de una enfermedad poco frecuente y poco sospechada, el equipo de salud no considera el nacimiento en un centro de tercer nivel ni las condiciones de recepción, destacando nuevamente la importancia de conocer dicha patología para obtener mejores resultados.
En el paciente que presentamos no planteamos diagnósticos diferenciales al contar con la confirmación prenatal9-11, en caso de no contar con ésta debemos tener presentes como diagnósticos diferenciales la epidermolisis ampollar hereditaria (EA) y el síndrome de piel escaldada estafilocócica12-14. Otros diferenciales son la necrolisis epidérmica tóxica, incontinencia pigmenti, infección herpética, pénfigo neonatal, hijos de pacientes con penfigoide gestacional, mastocitosis ampollar y ampollas traumáticas.
En cuanto a la recepción del recién nacido con piel frágil, se debe tener ciertas consideraciones al momento del nacimiento. En caso de contar con el diagnóstico prenatal, el nacimiento se recomienda realizarlo en un centro de tercer nivel que cuente con equipo multidisciplinario involucrando neonatólogos, dermatólogos, infectólogos, para abordar al paciente en su conjunto, verificar la presencia del kit con elementos imprescindibles para su recepción y así poder realizar un manejo acorde a su patología y prevenir complicaciones futuras.
El obstetra debe pautar la oportunidad para el nacimiento, no habiendo mayores contraindicaciones para el parto vaginal.
Nuestro paciente nació en el interior del país, donde fue recepcionado como un neonato sin patología, lo que implicó la estimulación por frotamiento aumentando las lesiones. Al momento del nacimiento los neonatos con piel frágil deben recepcionarse con campos estériles suaves, colocarlos en una termocuna sin humedad (esto favorece la producción de ampollas) evitando la hipotermia, sobre colchón con sábanas estériles. El clampeo de cordón debe realizarse con doble ligadura utilizando lino (no clips), no utilizar pulsera de identificación. El pañal se debe colocar recortando los elásticos evitando el roce y colocando vaselina estéril en los bordes. No se aconseja realizar el pasaje rutinario de sondas.
La ictiosis epidermolítica cursa con numerosas complicaciones, siendo las más graves en etapa neonatal la sepsis y el desequilibrio hidroelectrolítico. Nuestro paciente presentó una pustulosis a Staphylococcus aureus localizada, que respondió al tratamiento instaurado con buena evolución.
En cuanto al pronóstico, sabemos que la gravedad de esta enfermedad es variable, la evolución natural se caracteriza por un cambio de mecano ampollar a hiperqueratósico, que puede ser sectorial, más comúnmente en pliegues o generalizado. Esto se debe a que el mismo defecto de la barrera cutánea estimula la proliferación de queratinocitos, es decir, la necesidad de una barrera competente se vuelve primordial y se inician respuestas homeostáticas de reparación con inducción de hiperplasia epidérmica7. Podemos observar dicho cambio fenotípico en el control de nuestro paciente a los 7 meses de vida (Figura 3).
Esto tiene un impacto negativo en la calidad de vida, generando aislamiento social por el mal olor y aspecto cutáneo, prurito o infecciones, o ambos. Es una enfermedad que si bien no presenta compromiso sistémico, requiere tanto tratamiento tópico como sistémico, constante y crónico, lo que genera un gran impacto en la economía familiar12.
Los tratamientos tópicos incluyen emolientes, calcipotriol, liarozol y queratolíticos. El único tratamiento que mejora la dermatosis y permite lograr una calidad de vida aceptable son los retinoides, si bien deben mantenerse de por vida para lograr dicho cambio, reservándose para casos severos. El liarozol es un agente imidazol de la nueva clase de los agentes bloqueadores del metabolismo del ácido retinoico. Las formulaciones tópicas y orales de liarozol pueden brindar un mejor perfil de tolerabilidad en comparación con otros tratamientos, pero la ventaja del liarozol sobre la acitretina oral no está del todo aclarada y no se encuentra disponible comercialmente hasta la fecha3.
Estudios recientes han visto una mayor expresión de citoquinas de la vía Th17 relacionada con formas clínicas más severas de ictiosis, por tanto nuevos paradigmas terapéuticos de investigación están dirigidos a la vía IL-17/IL-23, siendo Secukinumab, un anticuerpo IgG1 monoclonal humano anti IL-17A, el que está en auge, aunque todavía en investigación13-16.
Conclusión
La ictiosis epidermolítica es una patología rara de curso crónico, que puede ser diagnosticada en forma prenatal o sospecharse si hay antecedentes familiares. Resulta de suma importancia planificar el momento del nacimiento así como contar con el kit de recepción para pacientes con piel frágil. Los cuidados estandarizados (pautas) mejoran el pronóstico y disminuyen las complicaciones en el período neonatal. Destacamos la importancia del abordaje de estos pacientes por un equipo multidisciplinario.

