











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
Las ganglionopatías sensitivas (o neuronopatías sensitivas) son una pequeña subcategoría de neuropatías periféricas, caracterizadas por la degeneración primaria del soma de la primera neurona sensitiva de los ganglios de la raíz dorsal y del ganglio trigémino. 1,2
Su etiología suele ser clasificada como hereditaria o adquirida. 3 Dentro de estas últimas, cobran especial importancia la paraneoplásica (siendo el carcinoma broncopulmonar el más vinculado), enfermedades autoinmunes (Síndrome de Sjögren fundamentalmente) e infecciones virales. 4
Dada su afectación característica en el sistema nervioso (SN), suelen dar una presentación clínica distintiva, dada por ataxia propioceptiva y alteraciones sensitivas en los miembros que aparecen de forma multifocal y asimétrica. 4
Es fundamental su diagnóstico temprano, ya que el tratamiento oportuno de la afección subyacente puede influir en el curso evolutivo, signando el pronóstico vital y funcional del paciente. 5
A continuación reportamos el caso de una paciente con diagnóstico de ganglionopatía sensitiva paraneoplásica, secundaria a un carcinoma broncopulmonar neuroendocrino a células grandes y realizamos una revisión narrativa de la literatura acerca del tema.
Caso clínico
Paciente de sexo femenino, 50 años con antecedentes personales de tabaquismo (índice paquete/año de 45).
Fue valorada en consulta ambulatoria con cuadro de 5 meses de evolución dado por parestesias en planta de pie y mano izquierdas, que luego se extendió al resto del miembro, asociando dolor de características neuropáticas en igual topografía.
Concomitantemente presentaba trastorno en la marcha progresivo, con marcada inestabilidad. Como fenómeno acompañante refería adelgazamiento de 15 kg, astenia y adinamia. No presentaba fiebre, dolor raquídeo, alteración deglutoria ni esfinteriana.
Al examen físico se evidenciaba bocio grado II, asimétrico a expensas de lóbulo izquierdo. En lo neurológico en miembros superiores las fuerzas estaban conservadas, hiporreflexia a izquierda, hipoestesia e hipopalestesia bilateral. En miembros inferiores fuerzas conservadas, hiporreflexia a izquierda con cutáneo plantar indiferente, hipoestesia e hipopalestesia a predominio distal. Presentaba marcha atáxica y presencia de Romberg sin latencia.
Inicialmente se plantearon dos posibles diagnósticos:
-Ganglionopatía sensitiva: Compromiso sensitivo exclusivo, inicio asimétrico y progresivo.
-Polineuropatía desmielinizante inflamatoria crónica (CIDP), variante sensitiva: Ataxia sensitiva sin compromiso motor, alejando este planteo la presentación asimétrica del cuadro.
En la tabla 1 se presentan los estudios dirigidos en vista a establecer un diagnóstico.

Con los estudios realizados, se confirmó el diagnóstico de Ganglionopatía sensitiva.
Se solicitaron estudios para su valoración etiológica: Serologías virales, anticuerpos para enfermedad autoinmune y búsqueda de neoplasia mediante diversos métodos de imagen y biopsia tiroidea (Tabla 2).
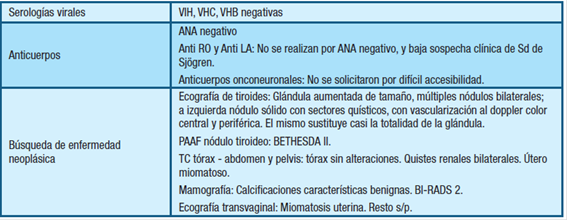
Se inició tratamiento con bolos de metilprednisolona 1 g i/v día por 3 días consecutivos. Dada la ausencia de mejoría, se realiza inmunoglobulina 1 g/kg día por 2 días consecutivos, sin cambios. Se realiza tratamiento de mantenimiento con prednisona 40 mg día, revalorando a la paciente en 15 días sin presentar mejoría.
Se reitera el estudio de conducción eléctrica 2 meses luego de instaurando el tratamiento, donde se evidencia trazado con mismas características que estudio previo, con leve progresión.
Dada la ausencia de beneficio se realiza descenso progresivo de prednisona hasta su suspensión.
Para completar el estudio etiológico y descartar la etiología paraneoplásica, se decide solicitud de PET-TC que informa: nódulo pulmonar hipermetabólico en lóbulo superior izquierdo (LSI), 13 mm diámetro, SUV 10, adenopatías hipermetabólicas aortopulmonar e hiliar ipsilaterales, 21 y 13 mm respectivamente, SUV 10 y 6. En suma: Nódulo pulmonar hipermetabólico LSI, en contexto clínico primer planteo carcinoma broncopulmonar (CBP), adenopatías de probable naturaleza secundaria.
La paciente fue valorada por cirugía de tórax, realizándose lobectomía superior izquierda con vaciamiento ganglionar mediastinal. La anatomía patológica informó Carcinoma neuroendocrino de células grandes, 4 ganglios linfáticos con compromiso metastásico. En suma: pT1b, pN1.
Evaluado el caso clínico en ateneo de oncología médica, se plantea escaso beneficio de adyuvancia quimioterápica, con eventual peoría de sintomatología neurológica por platinos, por lo que se mantiene con seguimiento clínico - paraclínico.
Se destaca durante toda la evolución de la enfermedad un muy difícil manejo del dolor neuropático. Es valorada en policlínica multidisciplinaria de dolor, con requerimiento de altas dosis de metadona (30 mg día) y coadyuvantes varios (duloxetina y gabapentina). Recibe sesiones de acupuntura, con escasa mejoría.
Al año de tratamiento quirúrgico, consulta en Hospital de Las Piedras por síndrome confusional. Se realiza Tomografía de Cráneo que evidencia múltiples procesos expansivos intraaxiales supratentoriales bilaterales, con edema perilesional y efecto de masa local, compatibles con secundarismo encefálico. Fallece a las 48 horas de dicho ingreso hospitalario.
Discusión
Las ganglionopatías sensitivas (GS) son un subgrupo poco frecuente de neuropatías periféricas.
Los ganglios sensitivos contienen los cuerpos celulares de los nervios sensoriales, encontrándose a ambos lados de la médula espinal, adheridos a la raíz dorsal de cada nervio espinal. Sus proyecciones proximales son las raíces nerviosas sensoriales que ingresan a la médula espinal y sus proyecciones periféricas son las fibras sensoriales de los nervios periféricos. 6 Las ganglionopatías sensitivas se caracterizan por dañar los cuerpos de las células nerviosas sensoriales de la raíz dorsal y los ganglios del trigémino, lo que lleva a la degeneración de sus proyecciones. 5
Dada la rareza de estas enfermedades, hay relativamente poca información publicada, siendo la mayoría de la bibliografía informes de casos aislados, series de casos retrospectivas o revisiones. 3
Estos trastornos pueden clasificarse en adquiridos, hereditarios o degenerativos. 3 En este reporte, nos centraremos en las de etiología adquirida, y su asociación con una enfermedad sistémica subyacente, generalmente inflamatoria, neoplásica o infecciosa, que a menudo se manifiesta después del desarrollo de los síntomas neurológicos. 6
Dentro de las causas adquiridas, encontramos las etiologías inmunomediada (paraneoplásica y no paraneoplásica), infecciosa, tóxica e idiopática (tabla 3). 4
Tabla 3: Tipos y causas de ganglionopatías sensoriales adquiridas* VEB - Virus de Epstein-Barr, VIH - virus de inmunodeficiencia humana, HTLV-1 - virus linfotrópico de células T humanas tipo 1, VZV - virus varicela zoster.
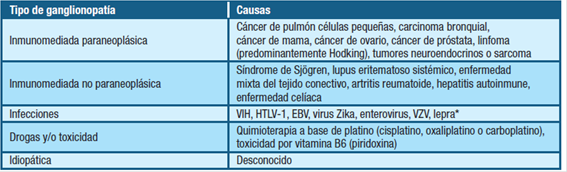
Es de destacar que en aproximadamente la mitad de los pacientes el trastorno sigue siendo idiopático, a pesar de una evaluación exhaustiva. 6
Los pacientes con GS paraneoplásica, inmunomediada, tóxica y posinfecciosa se presentan de forma subaguda. 4
Por estar en vinculación con el caso reportado, se profundizará acerca de la causa paraneoplásica. Las GS se encuentran entre los síndromes neurológicos paraneoplásicos más frecuentes. 4
La GS a menudo precede a los síntomas de la neoplasia y su diagnóstico, siendo la mediana de tiempo desde el inicio de la signo-sintomatología hasta el diagnóstico de cáncer de cinco meses, con variaciones de pocas semanas hasta cinco años. 5 Esto remarca la importancia de una búsqueda exhaustiva de una neoplasia maligna subyacente. 5
El cáncer de pulmón de células pequeñas es el tumor que se ha asociado con mayor frecuencia (cercana a un 80%) 7 aunque también se ha implicado otras neoplasias, dentro de las que se destacan los tumores neuroendocrinos. 5
Las manifestaciones clínicas dependen del tipo de neurona involucrada.
Las neuronas de fibras grandes median la propiocepción, por lo que su degeneración produce ataxia de la marcha. La lesión de neuronas de tamaño pequeño y mediano produce síntomas sensitivos irritativos: “dolor ardiente”, hiperestesia y alodinia. 4,5,8 En una serie de casos, el dolor fue una característica predominante en el 80% de los pacientes que recibieron este diagnóstico. 9 Cuando la pérdida de propiocepción es grave, el paciente puede presentar una marcada ataxia de la marcha y seudoatetosis de los dedos de manos y pies. También este déficit propioceptivo puede generar dificultad en el mantenimiento sostenido de la fuerza. 7 La afectación suele ser generalizada, multifocal, asimétrica y rápidamente progresiva. 4,5 La arreflexia ante una fuerza muscular conservada es invariable. 5
A menudo puede haber afectación de los ganglios autónomos, por lo que los pacientes también pueden tener disautonomía (por ejemplo hipotensión ortostática, arritmias cardíacas, dismotilidad gástrica y alteraciones de la sudoración). 6
La combinación de síntomas sensitivos proximales y pérdida sensorial con ataxia, con fuerzas conservadas, es el síndrome de GS arquetípico. 6
En 2009, Camdessanché et al. propusieron un conjunto de criterios diagnósticos para la GS sobre la base de una población de estudio retrospectiva, para distinguir la GS de otras neuropatías sensoriales 3,10. Estos criterios fueron validados en un estudio de 210 pacientes en 15 centros de referencia. En este estudio de validación, los criterios tuvieron una sensibilidad de 90.3% y especificidad de 85.2%, con un valor predictivo positivo del 91.9% y un valor predictivo negativo del 82.5%. 3,11
En la tabla 4 se presenta el formulario de puntuación para el diagnóstico de la revisión de GS de Kelly Graham Gwathmey. 3
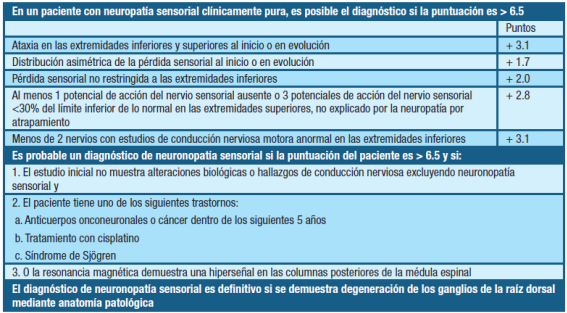
Los estudios paraclínicos se pueden dividir en dos grupos: los que confirman la presencia de una ganglionopatía sensorial, y los que buscan determinar la causa subyacente de la misma. 5
Para valorar presencia de GS se realizan estudios de electrodiagnóstico. Los estudios de conducción nerviosa sensorial típicamente demuestran una neuropatía sensorial no dependiente de la longitud con potenciales de acción de nervios sensoriales (SNAPs) difusamente disminuidos y eventualmente ausentes. En forma frecuente los miembros superiores se afectan en forma desproporcionada. 3 En estos casos, la resonancia magnética de la columna cervical puede mostrar hiperintensidad en T2 en las columnas dorsales de la médula espinal, debido al daño de las células ganglionares, con la consiguiente degeneración walleriana. Estos hallazgos demoran en desarrollarse, por lo que es posible no se vean en presentaciones subagudas. 5
Los estudios de conducción nerviosa motora suelen ser normales, pero puede haber reducciones subclínicas en el potencial de acción del músculo compuesto, en particular en pacientes con enfermedad paraneoplásica. 5
La biopsia de ganglio de raíz dorsal es el gold standard para la confirmación diagnóstica, pero no se recomienda su realización dado su carácter invasivo. 3
Para valorar etiología subyacente, el estudio diagnóstico incluye pruebas de autoanticuerpos séricos (antinucleares, anti-Ro y anti-La), tomografía de tórax y biopsia de masas sospechosas. 6 El PET-TC cobra relevancia en la búsqueda de neoplasia oculta, si los otros estudios imagenológicos fueron negativos. Si los estudios radiográficos de cribado (incluido PET-TC) son negativos, se recomienda repetir el cribado a los 3 o 6 meses y luego cada 6 meses durante al menos 4 años. 4
El examen del líquido cefalorraquídeo (LCR) en estos pacientes puede mostrar concentraciones elevadas de proteínas, pleocitosis leve o bandas oligoclonales. 6
Se han asociado dos anticuerpos con las ganglionopatías sensoriales paraneoplásicas: los anticuerpos anti-Hu (autoanticuerpos nucleares antineuronales tipo 1 - ANNA-1), y los anticuerpos contra la proteína mediadora de la respuesta a la colapsina-5 (CRMP-5). 5
Los anticuerpos anti-Hu son generados por el sistema inmunológico contra las células tumorales que expresan la proteína Hu, penetrando a nivel del ganglio de la raíz dorsal, donde la barrera hematoencefálica está fenestrada y es permeable. Sin embargo, el papel de los anticuerpos anti-Hu en el daño neuronal directo aún no se ha dilucidado. Se cree que el daño es secundario a respuesta de células T citotóxicas CD8. 3 Los estudios de autopsia han mostrado inflamación y pérdida de células ganglionares en los ganglios de la raíz dorsal y degeneración walleriana de las columnas posteriores en la médula espinal. 6
Estos anticuerpos tienen una alta especificidad (99%), pero baja sensibilidad (82%) y, por tanto, su ausencia no excluye una malignidad subyacente. Tampoco existe una correlación del título de anticuerpos con la progresión de la enfermedad. 5
Una serie de 170 pacientes con anticuerpos anti-Hu mostró que el 93% de los mismos tenían LCR anormal con un nivel medio de proteína de 78 mg/dL, y 43 de 73, de los pacientes con ganglionopatía sensitiva, tenían bandas oligoclonales. 4,12
En la tabla 5 se presenta la paraclínica a solicitar de la revisión de GS de Sarah Sheikh y Anthony Amato. 5

Una revisión recientemente publicada 6 recomienda considerar la solicitud de anticuerpos para enfermedad celíaca. También, en los casos de GS idiopáticas de fibras pequeñas y grandes, valorar realizar pruebas de anticuerpos anti-disacárido de heparina trisulfatada y anti-receptor 3 del factor de crecimiento de fibroblastos, ya que pueden encontrarse en pacientes con neuropatías o neuronopatías de predominio sensorial, aunque su especificidad y función patogénica no están claras. Para la evaluación de las GS idiopáticas de fibras pequeñas también podría considerarse la biopsias de piel de la parte distal de la pierna y el muslo, para medir la densidad de fibras nerviosas intraepidérmicas.
El tratamiento dependerá de la causa subyacente, por lo cual es fundamental su hallazgo oportuno, ya que mayores serán las probabilidades de mejora. 5
El manejo en la GS paraneoplásica es un desafío. La estrategia se basa principalmente en la opinión de expertos, ya que no se han realizado ensayos clínicos controlados aleatorios debido a la baja frecuencia de la enfermedad. 3
En un estudio retrospectivo, el tratamiento del tumor fue la única intervención que resultó en la estabilización del síndrome paraneoplásico en pacientes con anticuerpos anti-Hu, 3,13 aunque está descrito que generalmente no conduce a la remisión de la ganglionopatía. Es probable que la mejora con el tratamiento esté limitada por la destrucción del cuerpo celular de la neurona sensorial. 6
El segundo principio del manejo es el tratamiento inmunomodulador, realizándose terapia con corticosteroides (metilprednisolona) por vía intravenosa o con inmunoglobulina intravenosa mientras se evalúa la malignidad. Existe una ventana de oportunidad limitada para la estabilización de la enfermedad, ya que luego de la reacción inflamatoria, hay daño neuronal irreversible, donde las intervenciones terapéuticas probablemente sean poco efectivas. 3,14,15
En suma, se puede realizar alguna forma de inmunoterapia si el síndrome neurológico es progresivo y tiene una duración de días o semanas 6); sin embargo, una serie de casos mostró una mejoría en los pacientes que fueron tratados dentro de los primeros 2 meses después del inicio de los síntomas, con estabilización del trastorno en los tratados dentro de los primeros 8 meses. 6,14
Conclusiones
La presentación clínica y el estudio eléctrico característicos son fundamentales para el diagnóstico de esta afección.
La etiología más frecuente es la paraneoplásica, secundaria a CBP. En nuestro caso destacamos el tipo histológico. El carcinoma de pulmón neuroendocrino de células grandes es un subtipo histológico raro, de pronóstico similar al cáncer de pulmón de células pequeñas (15% aproximadamente de los CBP). 16
Al momento del diagnóstico de una probable afección paraneoplásica debemos recordar que en un considerable porcentaje de casos el tumor se evidencia tiempo después. De aquí la importancia del alta sospecha clínica y del seguimiento estrecho del paciente.
Cabe mencionar el gran impacto que tiene esta afección sobre la calidad de vida y que si bien en algunos casos se evidencia algún tipo de respuesta con el tratamiento oncológico, en otros no la hay, siendo muy limitadas las opciones terapéuticas y con pobres resultados.

