











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
El síndrome hemofagocítico (SHF), también conocido como linfohistiocitosis hemofagocítica, es una enfermedad infrecuente y grave, generalmente subdiagnosticada.
No se trata de una única enfermedad, sino de un síndrome clínico provocado por una respuesta inmunológica inadecuada a un desencadenante (infeccioso, neoplásico, reumatológico, tóxico o metabólico) que da lugar a un fenotipo inflamatorio característico. Esta respuesta inmune provoca una liberación desmedida o ¨tormenta de citocinas¨ responsables de las manifestaciones clínicas. 1)
En 1994, la Sociedad de Histiocitosis publicó el primer protocolo internacional de tratamiento y en 2004 se presentaron modificaciones con la información analizada durante 1994-2004, generando nuevos criterios diagnósticos y guías terapéuticas. Sin embargo, la variabilidad en la presentación clínica y la falta de especificidad en las pruebas de laboratorio hacen muy difícil y tardío el diagnóstico. 2) En pacientes portadores del virus de inmunodeficiencia humana (VIH), los desafíos diagnósticos y terapéuticos son aún mayores, lo que dificulta su abordaje.
El objetivo de este trabajo es revisar las distintas formas clínicas de presentación del SHF, destacando la importancia de una sospecha temprana a pesar de no cumplir totalmente con los criterios diagnósticos. Se comentan a continuación dos casos de SHF en pacientes portadores de VIH y se realizan las consideraciones diagnósticas y terapéuticas más relevantes.
Caso clínico 1
Hombre de 42 años, tabaquista desde los 16 años de edad (índice paquete/año de 10), con diagnóstico de VIH desde febrero 2019, en estadio C por Sarcoma de Kaposi cutáneo-mucoso y Pneumocitosis pulmonar (PCP). En tratamiento antirretroviral (TARV) con Dolutegravir y Lamivudina, profilaxis secundaria para PCP y profilaxis primaria para micobacterias atípicas.
Presentaba una historia de fiebre, hepatoesplenomegalia y pancitopenia leve, que se interpretó como un síndrome de reconstitución inmune (SIRI). Se realizó tratamiento empírico para Mycobacterium tuberculosis y para micobacterias atípicas, contando con un Ziehl-Nielsen de expectoración negativo y cultivos sin desarrollo. Linfocitos CD4 de 64 cel/mm, carga viral (CV) indetectable, evolucionando asintomático y sin citopenias durante 3 meses.
Re consulta por fiebre de 39°C axilar de 10 días de evolución acompañada de tos con expectoración mucopurulenta y repercusión general, agregando disnea a moderados esfuerzos. Al examen físico se encontraba lúcido, 25 rpm, febril y anemia clínica. Taquicardia de reposo de 110 cpm. A nivel pleuropulmonar estertores crepitantes bibasales y un síndrome en menos en tercio inferior de hemitórax izquierdo. Saturación de oxígeno ventilando al aire de 92% que corregía a 98% con oxigenoterapia. Abdomen indoloro, con hepatomegalia regular y esplenomegalia grado II.
Gasometría arterial: insuficiencia respiratoria severa tipo 1. Radiografía de tórax: infiltrado intersticio nodal a predominio basal, borramiento del ángulo costodiafragmático izquierdo. Analítica sanguínea: pancitopenia con hemoglobina de 7,4 g/dl, plaquetas de 77.000 cel/mm3, glóbulos blancos de 4000 cel/mm3, linfopenia de 900 cel/mm3 y 2300 cel/mm3 neutrófilos. Proteína C reactiva (PCR) de 136 mg/L y lactato deshidrogenasa (LDH) de 171 UI/L. Azoemia 0.35g/L Creatininemia 0.98mg/dl, hiponatremia leve de 127 mEq/l, tasa de protrombina (TP) 45%, y fibrinógeno 138 mg/dl. Bilirrubina total (BT) 1,12 mg/dl, bilirrubina indirecta 0,66 mg/dL, bilirrubina directa 0,44 mg/dL e hipoalbuminemia de 2,47 g/dl. Sideremia 17 ug/dl, transferrina 139 ug/dl, índice de saturación de transferrina 12% y ferritina 1378 ng/dl. Triglicéridos 500mg/dl.
Tomografía computada (TC) de tórax: imagen en vidrio deslustrado de distribución bilateral y difusa, derrame pleural bilateral de moderada entidad y adenomegalias mediastinales. TC de abdomen: hepatoesplenomegalia regulares, múltiples adenopatías mesentéricas y fina lámina de líquido subesplénico.
Estudio del líquido pleural: trasudado, citológico con escasos linfocitos. Cultivo del líquido pleural, de expectoración, hemocultivos, Ziehl-Nielsen y antígeno neumocóccico en orina negativos.
Con planteo diagnóstico de infección respiratoria baja subaguda con insuficiencia respiratoria severa y daño orgánico múltiple en paciente estadío SIDA, bajo TARV y con CV indetectable. Teniendo en cuenta la forma de presentación, los estudios de imagen y el nivel de inmunosupresión, se planteó etiología infecciosa, posiblemente secundaria a una micosis profunda. Como diagnóstico diferencial podía corresponder a una infección por Citomegalovirus (CMV) dado la agresividad en la forma de presentación. El Sarcoma de Kaposi pudo haber actuado como coadyuvante así como una enfermedad hemato-oncológica.
Se inició tratamiento empírico con ampicilina-sulbactam y trimetoprim-sulfametoxazol (TMP-SMX). No se encontró en oportunidad de realizar fibrobroncoscopía (FBC) por insuficiencia respiratoria severa.
Los cultivos de expectoración para micobacterias, hemocultivos para histoplasmosis, mielocultivo, examen de médula ósea para leishmaniasis, y antígeno criptococóccico, fueron negativos. Serologías para CMV, Parvovirus B19, virus Epstein-Barr (VEB), no reactivas. No obteniéndose diagnóstico etiológico.
La presentación clínica como enfermedad febril con afectación multiorgánica, en presencia de una hepatoesplenomegalia puso en sospecha la posibilidad de un SHF o un síndrome de overlap sepsis-SHF.
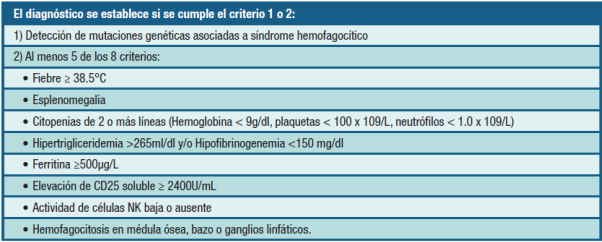
Presentó 5 de los 8 criterios diagnósticos establecidos para el SHF: fiebre, esplenomegalia, hemoglobina inferior a 9g/dl y plaquetas menores a 100.000/mm, hipofibrinemia e hipertrigliceridemia, y ferritina mayor a 500 ng/ml. El mielograma no evidenció hemofagocitosis, sabiendo que su ausencia no invalida el diagnóstico. 2-4
Se realizó tratamiento con anfotericina B, TMP-SMX, glucocorticoides intravenosos y transfusión de hemoderivados. Presentó una mala evolución con aumento progresivo y severo del derrame pleural, peoría de la pancitopenia, agregando insuficiencia renal aguda con hiperpotasemia severa, llevándolo al fallecimiento.
Caso clínico 2
Paciente de sexo masculino, 24 años, consumidor ocasional de cannabis. Comenzó 1 mes previo a la consulta con fiebre acompañada de disuria, polaquiuria y dolor abdominal inespecífico. Concomitante astenia y adinamia. Se trató de forma ambulatoria con cefuroxime, requiriendo posteriormente ingreso hospitalario dado la persistencia de fiebre.
Se obtienen serologías para VIH reactivas, carga viral de 80.000 copias/mL y CD4 de 28 cel/mm. La analítica al ingreso evidenciaba hemograma, creatininemia y azoemia normales. Fosfatasa Alcalina (FA) de 1200 UI/l, con bilirrubina y transaminasas normales. Urocultivos (UC) y hemocultivos (HC) persistentemente sin desarrollo. Ecografía de abdomen y aparato urinario: hepatoesplenomegalia, resto normal.
Evolucionó desfavorablemente persistiendo febril, con deposiciones líquidas, encefalopatía grado 3, peoría de la función hepática y colestasis (hiperbilirrubinemia (BT 35 mg/dl y TP disminuido). Agregó insuficiencia renal aguda con hiperpotasemia, acidosis metabólica, proteinuria y oligoanuria por lo que requirió hemodiálisis e ingreso a Unidad de Cuidados Intensivos.
Se completó valoración: serología para CMV y VEB, ambas, IgG positivo débil, IgM negativo. Serología para Hepatitis, Leptospira, Hantavirus, Brucellosis y Toxoplasmosis negativas. VDRL no reactivo. SARS-CoV-2 negativo. Galactomanano en sangre: 6.6 unidades (positivo). Anticuerpos antinucleares, antimúsculo liso y anti LKM negativos. Colangio-resonancia sin alteraciones.
TC de cráneo, cuello, tórax, abdomen y pelvis: hepatoesplenomegalia, sin adenomegalias. FBC con lavado bronquioloalveolar: reacción en cadena de la polimerasa para ADN de M. tuberculosis, baciloscopías y cultivos negativos, estudio para PCP negativo, galactomanano positivo: 2 unidades. Estudio del líquido cefalorraquídeo: sin alteraciones. Ecocardiograma transtorácico: normal.
Se hace el diagnóstico de micosis profunda, Aspergilosis invasiva, realizándose tratamiento con anfotericina B liposomal. Dado los síntomas digestivos e importante inmunosupresión en paciente grave, se sospecha probable coinfección por Mycobacterium Avium Complex, iniciándose de forma empírica azitromicina, moxifloxacina y etambutol.
Presenta mala evolución con anemia y plaquetopenia severa. Se plantea SHF secundario, por lo que se solicita ferritina (5.500 ng/dl) y triglicéridos (550 mg/dL). Se realiza mielograma que evidencia hemofagocitosis (Figura 1). Se inicia tratamiento con corticoides intravenosos. Agrega insuficiencia respiratoria y fallece a los 5 días.
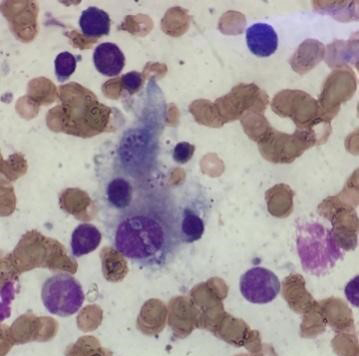
Discusión
Se presentaron dos pacientes con infección por VIH, con severa inmunodepresión, que se manifestaron con un cuadro grave en el que se arribó al diagnóstico de SHF secundario.
El SHF es una enfermedad infrecuente y de alta mortalidad. Fue descrita por primera vez en el año 1939 por Scott y Robb Smith. 5 Pertenece al grupo de las Histiocitosis. El término hemofagocitosis describe la presencia de macrófagos activados que incorporan dentro de su citoplasma eritrocitos, leucocitos y plaquetas. Cuando la hemofagocitosis ocurre en el contexto de una respuesta inmune exagerada e ineficaz, se denomina SHF. 1)
Se clasifica en SHF primario y secundario. El SHF primario se asocia a alteraciones genéticas específicas generalmente hereditarias y suele presentarse en la infancia, con una incidencia de 1 en 100.000 nacidos vivos en EE.UU. El SHF secundario se asocia a alguna enfermedad o condición subyacente y se presenta en la adultez. Sin embargo, hay casos reportados de adultos con SHF, en quienes se han determinado alteraciones genéticas de manifestación tardía. 6
El SHF se describió inicialmente vinculado a infecciones virales, pero posteriormente se ha relacionado a infecciones por hongos, bacterias y parásitos (particularmente, leishmaniasis).1 Una revisión de 2197 casos de adultos con SHF identificó que en el 50,5% el desencadenante fue una infección (predominantemente VEB y VIH), 48% malignidad, 44% en contexto de linfoma y 13% enfermedades autoinmunes, principalmente lupus eritematoso sistémico. 7-8 Cualquier estímulo intenso sobre la inmunidad celular puede desencadenar un SHF secundario. En contraste a la elevada incidencia de desencadenantes infecciosos vistos en el SHF pediátrico, el SHF del adulto es frecuentemente desencadenado por enfermedades hemato-oncológicas. 8)
En el paciente con VIH, el SHF puede ocurrir secundario a la infección por VIH, a infecciones oportunistas, como parte del SIRI o, incluso, por el propio TARV. Se reportó hemofagocitosis en el 20% de autopsias de 56 pacientes con VIH. 9-10 Un estudio retrospectivo de 58 pacientes que asociaban SHF y VIH encontró que los CD4 promedio eran de 91 cel/mm, 53% fue secundario a hemopatías malignas, 40% por infecciones, sobre todo tuberculosis y CMV, con una mortalidad promedio de 31%. 11 A nivel nacional, Dos Santos y cols, describen un caso de SHF asociado a VIH con inmunodepresión severa, CD4 de 10 cel/mm, en el que se realizó tratamiento etiológico empírico de infecciones oportunistas, corticoideo, transfusión de glóbulos rojos y factor estimulante de colonias de granulocitos. 12 Asimismo, en los casos analizados se constató inmunodepresión severa, 64 y 28 cel/mm respectivamente, y se planteó SHF secundario a infecciones oportunistas, realizándose tratamiento de las etiologías planteadas y específico del SHF, sin respuesta al mismo.
Existen criterios diagnósticos establecidos por la Histiocyte Society. 13-14 Se establece el diagnóstico con la presencia de 5 de los 8 criterios o con la detección de mutaciones genéticas asociadas al SHF. En los casos planteados se arribó al diagnóstico a través de la sospecha clínica, presentando 5 criterios diagnósticos. Tabla 1
La clínica es muy variada, pudiéndose presentar como fiebre de origen desconocido, hepatitis, falla hepática aguda, sepsis, enfermedad de Kawasaki o con síntomas neurológicos. 9 En el examen físico, la fiebre y la hepatoesplenomegalia son muy orientadoras, como se evidenció en los casos presentados.
A nivel paraclínico, se objetivan citopenias, hipertrigliceridemia y/o hipofibrinogenemia, e hiperferritinemia. Esta última puede alcanzar valores muy altos de entre 10.000 a 100.000 ng/dl. El hallazgo de hemofagocitosis en biopsia de médula ósea, bazo, ganglio linfático o hígado no es patognomónico de esta enfermedad y su ausencia no es excluyente, como se explicó previamente. 15
Los diagnósticos diferenciales son varios, siendo la sepsis la principal, como se planteó en el primer caso clínico. Otros son: la púrpura trombocitopénica trombótica, la coagulación intravascular diseminada, el síndrome hemolítico urémico, la encefalitis, el fallo hepático agudo y el síndrome de Dress.
La dificultad del diagnóstico se basa en distinguir entre la activación macrofágica fisiológica que puede haber en la sepsis, las enfermedades malignas o autoinmunes, y la activación patológica del SHF. Aunque los estudios microbiológicos confirmen una infección, esta puede ser la desencadenante del SHF, que causa un fallo multiorgánico progresivo, no respondiendo a la terapia antimicrobiana habitual, necesitando un tratamiento específico. 1
Debido a que el tratamiento puede ser esencial para la sobrevida y algunos de los criterios clínicos pueden aparecer tardíamente, no es necesario cumplir con todos los criterios para iniciar el mismo. 16 Se comenzará con las medidas de soporte necesarias, el tratamiento específico y el de la causa desencadenante, como se realizó en los dos casos clínicos comentados. Existe un protocolo internacional de tratamiento de la Histiocyte Society, basados en el uso durante 8 semanas de dexametasona, etopósido, ciclosporina y metrotexate intratecal más hidrocortisona (si existe compromiso neurológico), diseñado para el SHF primario, como también para cualquier forma severa de la enfermedad, para menores de 18 años. 13,14,17 Si bien este protocolo es utilizado también en adultos, no hay estudios sobre su eficacia. 7)
Si el paciente no presenta al menos una respuesta parcial en 2-3 semanas, se debe considerar terapia de rescate 9,17) con globulina antitimocito (GAT), infliximab (inhibidor de INF-gamma), alemtuzumab (anticuerpo monoclonal anti CD52), o trasplante alogénico. 11,16 Es primordial tratar simultáneamente la causa desencadenante para frenar el estímulo sobre la activación inmune. 17
El pronóstico es ominoso con una mortalidad mayor a 90% a 2 meses del diagnóstico. 6 Sin tratamiento la sobrevida es menor a dos meses.18) El linfoma como causa secundaria, la plaquetopenia, el aumento de la aspartato-aminotransferasa (ASAT) y de la LDH, y la edad mayor a 50 años son elementos de mal pronóstico. (17,18)
Conclusiones
Discutimos dos casos clínicos poco frecuentes y graves, en inmunodeprimidos, que se presentaron con fiebre y daño multiorgánico, realizándose el diagnóstico de SHF secundario. El SHF requiere de un alto grado de sospecha, por lo que siempre que estemos ante un cuadro de sepsis refractario al tratamiento, pancitopenia y visceromegalias, debemos considerar esta patología, para iniciar un tratamiento precoz y cambiar el pronóstico de esta enfermedad.

