











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
Las enfermedades por sobrecarga de hierro pueden ser primarias (congénitas) o adquiridas (secundarias). Las congénitas pueden asociarse a mutaciones en el gen HFE en hasta 95 % de los casos. Las secundarias se vinculan a otras enfermedades como son: las anemias con sobrecarga de hierro (por ejemplo, la talasemia mayor), la sobrecarga dietética de hierro, u otras enfermedades hepáticas (hepatitis C, esteatohepatitis no alcohólica), entre otras 1).
Se ha identificado una discrepancia entre la prevalencia genotípica y el número de casos diagnosticados en la clínica. Esto se explicaría tanto por el subdiagnóstico, como por factores que alteran la expresión fenotípica (sexo, edad, dieta), y por baja penetrancia genética 2).
La HH consiste en un grupo de trastornos genéticos caracterizados por un aumento en la absorción de hierro a nivel intestinal, con posterior acumulación del mismo en diversos tejidos 3). Entre ellos se destacan especialmente el corazón, hígado y páncreas. El compromiso de este último, vinculado al depósito de hierro en las células beta, se presenta clásicamente como una "diabetes bronceada". La importancia de la afectación hepática radica en que pasa desapercibida por largos períodos, por lo cual puede conducir a la cirrosis y posteriormente la aparición de hepatocarcinoma 4. Esto resulta en un diagnóstico tardío de una enfermedad potencialmente controlable, y puede conducir así a la muerte en un lapso de 5 a 10 años. Se presenta generalmente en varones de raza blanca, mayores de 50 años, alcanzando una prevalencia de hasta 0.5% en poblaciones europeas de origen nórdico. La mayor prevalencia en hombres se ha visto vinculada a la homocigosis C282Y del gen HFE como principal mutación vinculada con esta enfermedad 5).
La hepcidina es una hormona peptídica sintetizada en el hígado, constituyendo el principal regulador de la homeostasis del hierro corporal. Su concentración plasmática y distribución tisular regula la absorción del hierro a nivel intestinal, reciclaje por parte de los macrófagos y regulación de la eliminación del metal por medio de la ferroportina. Por ello, la ausencia de hepcidina por mutaciones en su gen codificador, parecerían constituir la causa final de la mayoría de las formas de hemocromatosis 6).
El tratamiento específico no resulta tan eficaz cuando el diagnóstico se hace en etapas tardías. Este consiste en depleccionar el hierro sérico, sea por flebotomías seriadas o donación regular de sangre 7).
En este artículo se presenta un caso clínico y se realiza una revisión de la literatura. Para la misma se consultaron las bases de datos Pubmed, Timbó y Sciencedirect con los términos de búsqueda “hemocromatosis hereditaria” y “HFE”, limitando la misma a artículos entre los años 2002 y 2015, exceptuando artículos clave buscados en forma dirigida.
Caso clínico
Se presenta el caso de un paciente de sexo masculino, de 50 años, con antecedentes personales de alcoholismo y con un diagnóstico reciente de Diabetes Mellitus. Presentaba antecedentes familiares de enfermedad hepática crónica de etiología incierta. Ingresó al servicio por hemorragia digestiva alta de entidad leve y autolimitada. Del examen al ingreso se destaca: regular estado general, fascies bronceado; palidez cutáneo-mucosa discreta; sin elementos clínicos de hiperestrogenismo. No presentaba al examen abdominal ascitis clínica ni visceromegalias.
Se confirmó endoscópicamente que la misma fue secundaria a una antro duodenitis erosiva crónica agudizada, si bien presentaba además una pequeña variz esofágica. De la paraclínica al ingreso se destaca: poliglobulia con hemoglobina de 17.6 g/dL, hematocrito de 51.2% y leucocitosis de 12740/mm3; plaquetas 175000/mm3. Del hepatograma se destacaba TGO 71 U/L, TGP 97 U/L, fosfatasa alcalina 168 U/L y GGT 48 U/L. El resto de la analítica básica se encontraba dentro de rangos de normalidad. Se solicitó una ecografía abdominal que informó hallazgos compatibles con hepatopatía crónica, sin elementos de hipertensión portal ni otras alteraciones a destacar. Se descarta por ELISA la presencia de infecciones por virus hepatotropos (virus de hepatitis B y C, virus de Epstein Barr, citomegalovirus).
Para estudio de la poliglobulia se solicitó un metabolismo férrico, destacando el ascenso marcado de la ferritina (5000 ng/mL), con una saturación de transferrina de 92%, sideremia de 226 µg/dL. Con planteo de sobrecarga férrica, se solicitó una RM abdominal con cuantificación del hierro hepático (Figura 1 ) que evidenció hallazgos compatibles con sobrecarga severa de hierro a nivel tisular.
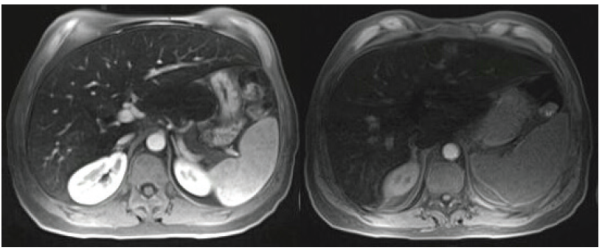
Figura 1: Resonancia abdominal. Se observa con (izquierda) y sin contraste (derecha) hipointensidad difusa a nivel hepático, compatible con sobrecarga férrica severa.
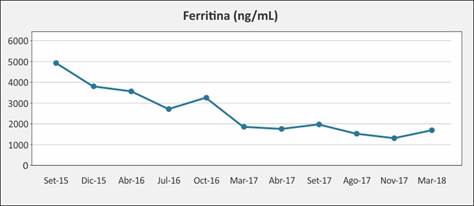
El diagnóstico de HH se confirmó mediante estudio genético que identificó la mutación homocigota del C282Y. Se inició tratamiento de inducción con flebotomías semanales, con evolución favorable. Adhiere además a Unidad de Trastornos Relacionados con el consumo de Alcohol (UNITRA), con cese del hábito tóxico. Si bien la adherencia al tratamiento con flebotomías fue irregular, el paciente presentó una mejoría lenta y paulatina del metabolismo férrico, como se muestra en la Gráfica 1.
Actualmente mantiene los controles ambulatorios, con controles clínicos y paraclínicos (hemograma y metabolismo férrico trimestral; ecografía abdominal y α-fetoproteína semestral). Se realizó también screening familiar de la HH por estudio genético de hijos
Discusión
a) Introducción
Se presentó la historia de un paciente con diagnóstico incidental de una cirrosis compensada de cuya búsqueda etiológica surgió la sospecha de HH. Afecta a una de hasta 400 personas de origen nórdico o céltico 8). Constituye uno de los principales trastornos hereditarios en caucásicos, siendo actualmente la enfermedad genética de transmisión autosómica recesiva más común, con penetrancia incompleta.
La variación de síntomas ocurre porque la condición de homocigoto para C282Y predispone al individuo a hemocromatosis. Usualmente se requieren factores individuales o ambientales adicionales para su expresión, como el abuso del alcohol en el caso de nuestro paciente 5).
Las principales causas de muerte en los pacientes con HH son el carcinoma hepatocelular (27%) y las complicaciones de la cirrosis (20%), seguidas por el cáncer extrahepático (12%), la miocardiopatía (7%) y el infarto de miocardio (6%). 3)
b) Historia
El síndrome clínico de diabetes, cirrosis e hiperpigmentación cutánea fue descrito inicialmente por Trousseau en 1865 “diabetes bronceada” 9). Pocos años después, en 1989, Von Recklinghausen adjudica el término de hemocromatosis para este síndrome, ya que identifica al hierro procedente de la hemólisis como pigmento localizado en los órganos afectados 10. En 1935,en una extensa serie de 311 casos, Sheldon determina el carácter hereditario de la hemocromatosis, que implica un error congénito del metabolismo que lleva a depósitos excesivos del hierro en diferentes tejidos 11. En los años setenta se describe la asociación genética con el complejo mayor de histocompatibilidad, localizando al gen responsable en el brazo corto del cromosoma 6 12. Veinte años después, Feder y su equipo descubren el gen HFE, y vinculan la mutación del mismo C282Y con la HH, hallazgo en 85% de los enfermos 13. Actualmente se plantea que más del 90% son homocigotos para C282Y, con una asociación menos clara para la mutación H63D 14.
c) Manifestaciones clínicas
Como se mencionó anteriormente, el cuadro clínico característico inicialmente descrito por Trousseau consistía en hombres de edad media, con diagnóstico de diabetes mellitus y tez bronceada. Si bien es un cuadro peculiar y muy orientador a la etiología, en la actualidad es poco frecuente, observándose en alrededor de 8% de los pacientes. Esto fue descrito por Adams y su equipo en los años 90 15, quienes plantean un cambio de escenario, lográndose en esa época un diagnóstico más precoz. Éste es en general incidental, ya que se detectan inicialmente síntomas inespecíficos, como son: artralgias, hepatomegalia, dolor abdominal, diabetes, entre otros. En otras oportunidades, se arriba al diagnóstico a partir de exámenes, guiado por la presencia de una ferritina sérica elevada, acompañada de un índice de saturación de transferrina también elevado. En el estudio de Adams se plantea un tiempo promedio de 5 años entre el inicio de los síntomas y la confirmación diagnóstica. Destacamos en nuestro paciente el abordaje diagnóstico tardío, dado que al momento de la consulta se presenta con compromiso hepático avanzado. Esto evidentemente conlleva implicancias terapéuticas y pronósticas.
En un estudio realizado en Madrid, publicado en el 2005, se intentó describir el fenotipo habitual de los pacientes con HH de la población asistida en el Servicio de Gastroenterología del Hospital Ramón y Cajal de esa ciudad 16). Se describió en esa oportunidad una predominancia de varones, con edad promedio de 47 años. Al momento del diagnóstico, la mayoría se presentaba con alguno de los siguientes síntomas: artralgias, hepatomegalia e hiperpigmentación cutánea. Casi un tercio de ellos presentaba diagnóstico de diabetes o insulinorresistencia.
En lo que respecta al Río de la Plata, no contamos verdaderamente con estudios en nuestro país; la hemocromatosis está incluida dentro de las enfermedades raras. En cuanto a Argentina, en 2017 fue publicado un estudio que estudia la sobrecarga de hierro en la dieta de varones con hemocromatosis 17; en este país tampoco se conoce la incidencia real de la enfermedad.
Se ha identificado que en mujeres el cuadro aparece de forma más tardía y menos grave, posiblemente vinculado a un efecto protector de la menstruación y el embarazo 2.
d) Estudios complementarios
Metabolismo férrico
En la práctica clínica, se continúa utilizando la ferritina como principal parámetro de monitorización de sobrecarga férrica 18. Esto se debe a que la sobrecarga de hierro genera aumento de producción hepática y liberación de ferritina a la sangre. Necesita niveles altos de hierro circulante para elevarse, por lo cual resulta menos sensible y específico que otros parámetros como el índice de saturación de transferrina (IST).
Sus niveles por encima de 400 ng/mL en hombres, y de 300 ng/mL en mujeres, aumenta la sospecha diagnóstica. Debe considerarse el hecho de que la ferritina constituye un reactante de fase aguda, por lo que puede verse alterado en otras circunstancias.
Por otra parte, la elevación del IST es la manifestación paraclínica más temprana de la enfermedad, constituyendo un punto habitual de partida para el diagnóstico precoz de la enfermedad. Valores de al menos 60% en hombres y 50% en mujeres nos permite detectar casi 90% de los enfermos homocigotos, siendo el punto de corte 45%.
Resonancia magnética
En la actualidad, la resonancia magnética (RM) es el mejor método no invasivo para determinación de sobrecarga férrica, ya que el 70% del hierro se almacena en el hígado 19). La concentración hepática de hierro (CHH) es menor de 36 mol/g en condiciones normales, ascendiendo frecuentemente por encima de 80 mol/g en las hemocromatosis.
La RM detecta la sobrecarga férrica debido al efecto paramagnético del hierro, es decir que el campo magnético de la técnica genera alineación paralela del hierro. Esto resulta en un acortamiento del tiempo de relajación T2, con disminución de la señal proporcional a la CHH. Por ello, la resonancia permite estimar la sobrecarga férrica hepática por dos métodos:
a) Cálculo de tiempo de relajación: es el más directo y más correcto. Se mide el tiempo requerido para que la magnetización genere un acortamiento del 37% de la medición inicial. A ese valor de T2 se le puede aplicar fórmulas matemáticas para estimar de manera confiable la CHH en mmol/g.
b) Medida del ratio de intensidad de señal: mide relación entre intensidad de señal hepática y de musculatura paravertebral. Esta última es usada como referencia ya que no se ve afectada por sobrecarga férrica en la hemocromatosis. La sobrecarga de hierro en el hígado se traduce en reducción del ratio hígado/músculo. Este método tiene como ventajas la sencillez de ejecución y la accesibilidad.
En lo que respecta a afectación cardíaca, se toma como punto de corte habitualmente ferritina 1000 mg/L para estudio de los depósitos en este órgano. En estos se solicita cardiorresonancia con secuencia T2*, que implica el uso de secuencias en eco. En base a este estudio, se clasifica a los pacientes en tres grupos:
Aunque la recomendación general es de resonancia anual, en los pacientes considerados “verdes”, con buena función cardíaca y buena adherencia al tratamiento, podrían espaciarse cada 2 o 3 años. Por el contrario, en los pacientes “rojos”, con función cardíaca reducida o interrupción previa del tratamiento, debe considerarse la realización de cardiorresonancia semestral.
La resonancia cardíaca permite valorar la función cardiaca, se ha aceptado que niveles bajos de T2* conllevan elevado riesgo de afectación miocárdica, aún si la función ventricular está conservada.
En lo que respecta a la sobrecarga férrica pancreática, aún no se ha definido correctamente el uso de la RM para determinarla.
Biopsia hepática
No es un estudio a solicitar de rutina, sino que se adapta según el caso; en nuestro paciente, dada la presentación clínica y genotípica, resulta innecesaria. Se justifica únicamente en casos de sospecha de cirrosis, con un fin meramente pronóstico 20). En este sentido, puede incluso en algunos pacientes recurrirse al FibroScan ®, que resulta menos invasivo y aporta similar información en cuanto a grado de fibrosis hepática.
En el eventual caso que se acceda a la biopsia, el anatomo-patólogo debe ponerse como objetivos: 1) determinar la presencia de sobrecarga férrica a nivel hepatocelular, 2) definir la presencia de cirrosis, y 3) descartar la presencia de enfermedades concomitantes.
En la HH, el hierro se deposita como hemosiderina; esto ocurre más intensamente a nivel periportal que a nivel centrolobulillar. En fases más avanzadas, puede observarse depósito férrico en las células de Kupffer y en los macrófagos portales, procedente del hierro liberado por los hepatocitos necrosados. Cuando el hierro se localiza exclusiva o preferentemente en macrófagos, la hemocromatosis probablemente sea secundaria. En cambio, cuando el depósito de hemosiderina hepatocelular es leve y prácticamente exclusivo de las áreas periportales, resulta compatible con fases iniciales de hemocromatosis o se trata de otra causa de hemosiderosis (por ejemplo, porfiria cutánea tarda). En la mayoría de los casos de hemocromatosis, existe un grado variable de fibrosis; resulta importante definir si es compatible o no con etapa cirrótica. Por último, interesa destacar que focos sin hierro evidencian transformación neoplásica, y obligan a un seguimiento estrecho del paciente.
e) Confirmación diagnóstica
Para confirmar el diagnóstico de HH debemos recurrir al estudio genético, con el objetivo de buscar las principales mutaciones del gen HFE conocidas como responsables. La prueba confirmatoria para el diagnóstico de hemocromatosis tipo 1 es el estudio del gen HFE (disponible en casi todos los laboratorios), pudiendo ser C282Y/C282Y, C282Y/H63D o C282Y/ S65C 20,21. La evaluación genética de la hemocromatosis no HFE (genes HJV, HAMP, TFR2 y SCL40A1) solo se realiza en unos pocos centros en el mundo 1.
La más habitual y conocida es la mutación C282Y, consistente en la sustitución de una cisteína por una tirosina en la posición 282. Otra mutación ampliamente conocida es la H63D, en la cual una histidina de la posición 63 es sustituida por un ácido aspártico. Ninguna de estas dos mutaciones en estado heterocigoto parece producir la enfermedad por sí sola, aunque se ha descrito un estado heterocigoto compuesto que puede acompañarse de sobrecarga férrica de grado moderado, y que se ve en un 2-8% de los pacientes con HH.
f)Tratamiento
Existen escasos avances en lo que respecta al tratamiento. A destacar del pilar higiénico-dietético, se considera de vital importancia el abandono de hábitos tóxicos (especialmente consumo de alcohol).
Interesa además aconsejar el consumo de alimentos que bajen la biodisponibilidad y absorción de hierro a nivel intestinal. Se deberá limitar también el consumo de mariscos crudos para evitar el riesgo de infección por Vibrio vulnificus, favorecida por la presencia de elevado contenido de hierro tisular. Tampoco debería consumirse vitamina C en demasía dado que favorece la absorción de hierro 20.
El pilar fundamental lo constituye la realización de flebotomías seriadas de 400 a 500 mL, semanal o quincenal. Esto constituye la etapa de inducción, que se mantiene hasta que la ferritina plasmática descienda por debajo de 50 ng/mL. Luego, las flebotomías se espacían 3-4 meses, previa solicitud de hemograma, de forma que el hematocrito no baje más de 20% del valor inicial1. De esta forma se logra depleccionar el exceso férrico, ya que con cada flebotomía se extraen entre 200 y 250 mg de hierro, dependiendo de la concentración de hemoglobina. La combinación de flebotomías con eritropoyetina acelera la depleción férrica.
Como alternativa a las flebotomías pueden utilizarse los quelantes del hierro, que se reservan para los pacientes intolerantes a las flebotomías 22. Se ha descrito que el uso de deferasirox a dosis de 5-15 mg/kg/día vía oral resulta eficaz, aunque debido a la presencia de efectos adversos reversibles pero no despreciables, y su costo, no ha sido muy difundido su uso. Dado que nuestro paciente se encuentra en etapa de cirrosis, se desaconseja el uso de este medicamento, dado que puede generar descenso en la albumina y recuento plaquetario.
Menos frecuentemente se practica la eritrocitoaféresis isovolémica, en general con previa administración de eritropoyetina. Esto consiste en la extracción de glóbulos rojos luego de estimular su producción, lo cual llevaría a una rápida depleción del contenido férrico circulante. En general tiene buena respuesta y pocos efectos adversos, pero debido a la complejidad de la técnica y sus costos, prácticamente no se utiliza en nuestro medio. Se describen trabajos con eritroaferesis de gran volumen, cuya ventaja es espaciar las venopunciones, haciendo el procedimiento más tolerable.
Los controles regulares de respuesta al tratamiento consisten en solicitud de hemograma y metabolismo férrico de forma trimestral. El objetivo del tratamiento consiste en alcanzar niveles de ferritina de entre 50 y 100 µg/L. cabe destacar, como se realizó con nuestro paciente, la importancia del screening de hepatocarcinoma con ecografía abdominal y α-fetoproteína semestrales.
Se aconseja además la búsqueda de hemocromatosis en familiares de primer grado, inicialmente con estudio genético. De presentar las mutaciones antes mencionadas, se solicita metabolismo férrico para determinar la presencia o no de la enfermedad, dado que como se mencionó, se trata de una enfermedad con penetrancia incompleta (21,22).
Conclusiones
Se reportó un caso de HH de diagnóstico incidental en la edad adulta. Afección poco común en nuestro medio, siendo infradiagnosticada dado que se manifiesta de forma tardía. Esta enfermedad, aunque descrita hace muchos años, ha presentado pocos avances respecto a su manejo terapéutico, posicionando a las flebotomías como el único tratamiento eficaz probado hasta el momento, como se practicó en el caso presentado.
El hallazgo incidental de esta patología en nuestro paciente - en etapa de cirrosis - concuerda con lo descrito en la literatura, donde trabajos muestran que en el 81% de los pacientes homocigotos para C282Y, la combinación de ferritina >1000 μg/l, elevación de AST (>40 U/l) y plaquetas <200 000/mm3 se asocia a la presencia de cirrosis 23.

