











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
PermalinkIntroducción
El síndrome de Li-Fraumeni (SLF) es una enfermedad hereditaria autosómica dominante con elevada penetrancia.
Es una enfermedad fenotípicamente y genéticamente heterogénea que se caracteriza por la aparición precoz de múltiples tumores en un individuo y en varios miembros de su familia 1), existiendo una correlación genotipo/fenotipo. Los tumores más frecuentemente implicados en este síndrome son: sarcomas de partes blandas y osteosarcomas, cáncer de mama (CM) de inicio temprano, tumores cerebrales, leucemias y carcinomas suprarrenales.
Existen dos criterios clínicos de sospecha: SLF clásico 2 y Li-Fraumeni Like (LFL) que comparte algunas características con el SLF, pero no todas 3).
Aproximadamente el 70% de los pacientes que cumplen criterios clínicos para su diagnóstico son portadores de una mutación de línea germinal en el gen TP53 localizado en el cromosoma 17p13. El gen TP53 es un supresor tumoral que cumple una importante función en el control de la estabilidad genómica. El producto del gen, la proteína p53, puede retrasar la progresión del ciclo celular, lo que permite una oportunidad para la reparación del ADN o la iniciación de la muerte celular programada (apoptosis). En ausencia de la proteína, las células que contienen el ADN dañado pueden sobrevivir y proliferar, lo que contribuye a acumulación de mutaciones y en consecuencia a la transformación maligna. Una variedad de técnicas moleculares se han utilizado para detectar mutaciones en el gen p53, incluyendo secuenciación de nueva generación, secuenciación tradicional por el método de Sanger y detección de grandes rearreglos, que evalúan la totalidad del gen y una región específica respectivamente. Se han identificado más de 300 mutaciones puntuales, de tipo sin sentido, distribuidas en regiones específicas del gen (hot spots), más frecuentemente entre los exones 5 a 8, si bien pueden detectarse, aunque con menos frecuencia, variantes patogénicas de cualquier tipo 4).
En los portadores de una mutación patogénica, se estima que el riesgo de desarrollar cáncer es del 50 % a los 31 años de edad para las mujeres y a los 46 años para los hombres siendo cerca del 100 % para ambos sexos a los 70 años. Los varones portadores de una mutación germinal parecen tener un riesgo similar al de las mujeres en forma más tardía, lo que estaría vinculado a la presencia de tumores ginecológicos. El riesgo se iguala en ambos sexos a mayor edad 5). El riesgo de desarrollar un segundo cáncer es alto, especialmente un cáncer inducido por radiación 6). Recientemente se han postulado mutaciones en células germinales de familias con SLF en otros genes como el CHEK2 que actúan en la ruta de la regulación del ciclo celular del TP53 en respuesta al daño del ADN aunque bien su implicación clínica no ha podido ser todavía claramente establecida 7).
En Uruguay no hay reportes publicados de grupos familiares portadores de la mutación hasta la fecha. Presentamos una paciente con diagnóstico de SLF que fue asistida en el Servicio de Oncología del Hospital de Clínicas y revisión de la literatura en PubMed desde 2000-2016
Caso Clínico
Paciente sexo femenino de 31 años, fumadora. Con antecedentes familiares de madre y dos tías maternas diagnosticadas de cáncer mama (CA) antes de los 40 años, prima portadora de la mutación germinal de p53, diagnosticada de rabdomiosarcoma en la infancia y de CM a los 31 años.
Consultó por disconfort a nivel pélvico, se realizó ecografía ginecológica constatándose una tumoración con epicentro pelviano, de 15 cm aproximadamente, la cual no impresiona ser de origen ginecológico. Con el planteo de un tumor de partes blandas pelviano no metastásico se realiza la resección de la masa pelviana macroscopicamente completa. La anatomía patológica evidencia un condrosarcoma grado 2 de 100x100x82 mm con márgenes libres y suficientes. Se realiza resonancia nuclear magnética (RNM) abdomen y pelvis postoperatoria que no evidencia lesiones por lo que permanece en controles clínicos e imagenológicos. A los 8 meses se constata en RNM mamaria de control un realce tipo no masa, de distribución segmentaria, homogéneo, BIRADS 4, sin traducción ecográfica y en la RNM abdomen y pelvis se evidencia recaída a nivel pelviano. Se realiza tomografía (TC) de tórax que no evidencia lesiones metastásicas y una core biopsia de lesión mamaria que evidencia un carcinoma ductal infiltrante (CDI) de mama tipo NOS, grado histológico final (GHF) 2, score 6. Receptor de estrógeno (RE) + 98%, receptor de progesterona (RP)+ 98 %, receptor del factor de crecimiento epidérmico-2 (HER2): 2 cruces, FISH no amplificado. Ki 30-40%.
La paciente realizó el estudio genético a través de la policlínica de oncogenética del Grupo Colaborativo Uruguayo (Hospital Central de las Fuerzas Armadas), dedicado al diagnóstico e investigación de enfermedades oncológicas hereditarias. Se determina a través de secuenciación por el método de Sanger su estado de portadora de la mutación familiar ya conocida 375G>C en el exón 4 del gen TP53. Se realizó el estudio genético a sus dos hijos siendo negativo en ambos casos.
Se discutió el caso en ateneo multidisciplinario y se resolvió realizar cirugía de las masas pelvianas, no siendo posible la resección macroscópicamente completa. La anatomía patológica confirma una recidiva de condrosarcoma grado 2. Valorado en ateneo de oncología médica y con el planteo de recaída de condrosarcoma pelviano resecado en forma incompleta y de CM T2N1MO RE y RP + HER2 2 negativo se decide realizar tratamiento sistémico paliativo en base a adriamicina y ciclofosfamida, siendo la adriamicina una droga activa tanto en el CM como en el condrosarcoma, mientras que la ciclofosfamida es activa únicamente en el CM. La paciente recibe 3 ciclos de quimioterapia con buena tolerancia y estabilidad lesional a nivel mamario pero con progresión lesional a nivel pelviano por lo que se inicia tratamiento para el CM en base a tamoxifeno asociado a goserelina.
Discusión y comentarios
En 1969 Frederick Li y Joseph Fraumeni describieron por primera vez el síndrome luego de identificar a 4 niños con sarcomas que eran parientes de primer grado de otros niños diagnosticados de rabdomiosarcomas 8).
Posteriormente en 1988 identificaron a 24 familias en las que 151 de sus integrantes habían sido diagnosticados de cáncer 2). La mayoría habían sido diagnosticados de sarcomas óseos o de partes blandas y CM y aproximadamente el 80 % antes de los 45 años, como en el caso de nuestra paciente. Otros tumores que evidenciaron con mayor frecuencia fueron: tumores cerebrales, leucemia y carcinoma de la corteza suprarrenal. Estudios más recientes han confirmado estos hallazgos 9,10).
En 1990 se identificaron las mutaciones en la línea germinal del gen p53 en las familias portadoras de SLF. Veinticuatro años más tarde, a pesar de las actuales controversias, el único gen que ha demostrado estar involucrado en el SLF es TP53 11,12).
La mutación identificada en nuestra paciente fue la 375G>C en el exón 4 de TP53 que genera una variante sinónima, (el cambio de base en el codón mantiene el mismo aminoácido,Thr) y afecta un sitio de "splicing" .
El espectro de tumores que se incluyen en el SLF es diverso e incluye: sarcomas de partes blandos y osteosarcomas, CM en la premenopausia, tumores cerebrales, leucemia y carcinomas suprarrenales. Otros tumores que tienen mayor prevalencia entre los pacientes portadores de la enfermedad son los melanomas, tumores de células germinales, tumor de Wilms, carcinomas gástricos, colónicos, pancreáticos y de pulmón (Tabla 1).

Al igual que con los tumores malignos comúnmente asociados con el SLF, estos también se diagnostican a una edad mucho más temprana que lo que ocurre en pacientes sin una mutación TP53. Una vez que la enfermedad se desarrolla, su comportamiento es en general similar al de los pacientes sin SLF, exceptuado por el diagnóstico realizado a una edad más temprana. Sin embargo, la susceptibilidad a un segundo tumor primario y las repercusiones del diagnóstico en el resto de la familia son factores importantes en el manejo de estos pacientes. Se estima un 50% de probabilidad de padecer un nuevo tumor maligno durante los primeros 10 años. Los individuos afectados tienen mayor riesgo de desarrollar cáncer en las diferentes etapas de su vida, en lo infancia: sarcomas (osteosarcomas y sarcomas de partes blandas), tumores cerebrales y tumores adrenocorticales y en edad adulta: CM en la premenopausia y sarcoma de partes blandas. La mitad de los portadores desarrollarán cáncer antes de los 30 años y un 15%, 4% y 2% de los individuos desarrollan 2, 3 y 4 tumores, respectivamente 10,13).
El CM es frecuentemente RE/RP+ y HER2 negativo. La frecuencia de tumores RE/RP+ es similar al del CM no asociado a mutación del gen p53 mientras que la frecuencia de HER 2 positivo es superior (20% vs 80%) 10,14). Sin embargo el tumor de nuestra paciente fue RE/RP+ HER2 2 cruces, FISH no amplificado. La heterogenicidad debido a la gran variedad de tumores que pueden aparecer en estos pacientes hace que sea difícil realizar un diagnóstico de SLF.
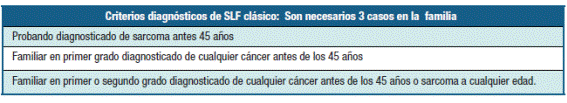
Existen criterios diagnósticos clínicos para el SLF descriptos por Li y Fraumeni en 1969 (Tabla 2), llamados criterios diagnostico SFL clásicos 8 y otros menos restrictivos descritos por Birch et al en 1994 3 (Tabla 3).

En el 2009 el Grupo de Trabajo Francés para el SLF describió los criterios de Chompret que incluyen las tres situaciones clínicas sugestivas de SLF: presentación familiar, los tumores primarios múltiples y los cánceres raros 15,16 (Tabla 4).

Estos 3 criterios diagnósticos fueron comparados en 525 pacientes derivados para el estudio de la mutación de TP53 9). Se identificaron mutaciones en 91 de 525 casos (17 %). Los criterios de SLF clásicos y los criterios Chompret fueron los de mayor utilidad. Cuando se combinaron estos criterios, se detectaron 71 de 75 familias con mutaciones del gen p53 (sensibilidad del 95% y especificidad de 52%). El diagnóstico clínico de SLF clásico o LFL debe ser confirmado por la presencia de una mutación del gen p53. En los exones 5, 6, 7 y 8 se localizan el 80% de las mutaciones responsables de SLF. Una vez confirmada la mutación en el probando se puede estudiar a los familiares con sospecha diagnostica ya que si bien el diagnóstico precoz de la enfermedad no predice la edad de inicio, la severidad ni la progresión de la mismo, es posible realizar estudios de screening que permitan llegar a un diagnóstico precoz.
Actualmente no existen guías de tratamiento oncológico específicas para estos pacientes, por lo que su tratamiento es similar al de los pacientes sin mutación p53, con la excepción de minimizar la exposición a radiación debido a que se han reportado tumores malignos en zonas irradiadas lo que sugeriría una carcinogénesis postrádica. Por esta razón y teniendo en cuenta que el riesgo de desarrollar CM es superior al 50 % a los 40 años de edad, es que cuando estas pacientes son diagnosticas de CM se prefiere la mastectomía en lugar de la tumorectomía seguida de radioterapia 13,17,18).
En el seguimiento de estos pacientes se debe tener en cuenta sus antecedentes personales y familiares. En la actualidad se encuentran disponibles diferentes pautas de seguimiento para estos pacientes 18-20). Se recomienda realizar en forma periódica un riguroso examen de la piel, un examen neurológico y evitar la exposición a agentes carcinogénicos tales como el tabaquismo y las radiaciones ionizantes. Con respecto al CM las recomendaciones incluyen el autoexamen mamario mensual y realizado por un médico dos veces al año a partir de los 18-20 años y controles imagenológicos anuales a partir de los 20 o 25 años (mamografía o resonancia magnética). Para la determinación temprana de los sarcomas se recomienda un RNM de cuerpo entero en forma anual.
Si bien aun un no se ha logrado definir cuál es la mejor estrategia de detección precoz, un estudio que evaluó el papel del screening en estos pacientes a través de RNM y parámetros bioquímicos se evidencio un beneficio en sobrevida global a 5 años para los pacientes sometidos a vigilancia (88,5% vs 59,6%) 21,22), sin embargo son necesarios más estudios para conocer el real beneficio la RNM en estos pacientes.
Conclusiones
El SLF es una enfermedad hereditaria autosómica dominante con elevada penetrancia, que se caracteriza por la aparición precoz de múltiples tumores en un individuo y una marcada agregación familiar. Presentamos el caso de una paciente de 31 años a la que se diagnostica un condrosarcoma pelviano y posteriormente un CM localizado. Se identifico la mutación 375G>C en el gen TP53 mediante secuenciación Sanger. Actualmente no existen guías de tratamiento específicas para el SLF, por lo que su tratamiento es similar al de los pacientes sin mutación, minimizando la exposición a la radioterapia debido a reportes de tumores malignos en zonas irradiadas. Sin embargo, la suceptibilidad a un segundo tumor primario y las repercusiones del diagnóstico en el resto de la familia son factores importantes en el manejo de estos pacientes. En la actualidad se encuentran disponibles diferentes pautas de seguimiento para estos pacientes. Se recomienda realizar en forma periódica un riguroso examen de la piel, un examen neurológico y evitar la exposición a agentes carcinogénicos tales como el tabaquismo y las radiaciones ionizantes. Con respecto al CM las recomendaciones incluyen el autoexamen mamario mensual y realizado por un médico dos veces al año a partir de los 18-20 años y controles imagenológicos anuales a partir de los 20 o 25 años (mamografía o resonancia magnética). Para la determinación temprana de los sarcomas se recomienda un RNM de cuerpo entero en forma anual.

