











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
La Retinosquisis ligada al cromosoma X es una enfermedad retiniana degenerativa relativamente frecuente, de comienzo en edad temprana, que afecta principalmente al sexo masculino, aunque se han visto casos excepcionales de niñas portadoras con pequeñas anomalías de retina.1
La forma de herencia es ligada al cromosoma X recesiva, presenta penetrancia completa y expresividad variable, y no se ha determinado una correlación genotipo-fenotipo definitiva.
Si bien esta enfermedad fue descripta por el oftalmólogo austríaco Josef Hass en 1898, el gen responsable, RS1 (Xp22.13), fue identificado recién en 1997 por el equipo de Sauer et al. 2 Este gen posee 6 exones y codifica una proteína extracelular de 24 Kda altamente conservada. Se expresa exclusivamente en fotorreceptores y células bipolares de la retina, como una proteína de superficie celular que participa en las uniones célula-célula, siendo esencial para mantener la integridad funcional y estructural de la misma. (3
Al momento se conocen cerca de 190 mutaciones patogénicas en este gen: 80% son mutaciones puntuales, 13% deleciones, 3% duplicaciones, 1.5% inserciones y otro 1.5% indels. Un 40% de ellas causan un codon de stop prematuro o un cambio en el marco de lectura de la secuencia, lo que lleva a producir una proteína no funcional. Esto hace pensar que el principal mecanismo patogénico es la pérdida de función del producto del gen. (3
La prevalencia de esta enfermedad ronda entre 1:5000 a 1:20000 varones en todo el mundo; no contamos con registros epidemiológicos en nuestro país.4
A nivel clínico se caracteriza por pérdida moderada a severa de la visión central, provocados por episodios similares a desprendimiento de retina, focales, en forma de quistes llamados esquisis, que suelen revertir de forma espontánea. En ocasiones presentan complicaciones importantes como son la hemorragia intraocular o el desprendimiento de retina propiamente dicho.
Inicia en la primera infancia, presenta luego un período de estabilidad para, cerca de la tercera a cuarta década de la vida, iniciar un deterioro progresivo. El fondo de ojo puede ser de utilidad viéndose cambios microquísticos en la región macular acompañados en oportunidades de velos vítreos. (5
El hallazgo de la mutación causante de la patología es de gran importancia tanto para el paciente, al que le permitiría arribar al diagnóstico genético, como para sus familiares, en quienes podría determinarse el estado de portador, y realizar asesoramiento e incluso prevención.
Se presenta un caso ilustrativo en quien se logró establecer un diagnóstico clínico altamente presuntivo, el cual se confirmó a nivel molecular mostrando la segregación de una mutación en RS1 a través de varias generaciones de la familia. La variante en RS1 encontrada en este paciente solo ha sido descripta a nivel mundial en una familia de origen Chino con varones afectados de Retinosquisis, por lo que encontrar este caso apoyaría la patogenicidad de la misma.
Caso clínico
Paciente de 2 años, sexo masculino, hijo de pareja sana no consanguínea. Sin patología perinatal evidente, bien controlado en salud, con buen crecimiento y desarrollo. Sin dismorfias.
A los 20 meses de vida sus padres detectan episodios que impresionan de pérdida de visión (ej.: chocarse con muebles, no lograr tomar objetos), además de exodesviación de ojo izquierdo, por lo que es valorado por Oftalmólogo, quien solicita de forma urgente tomografía de órbita por probable desprendimiento de retina en ojo izquierdo.
En la anamnesis surge, además, el dato de familiares varones por línea materna con patología ocular congénita ligada al cromosoma X.
Se ingresa al paciente al área de internación de pediatría de su prestador de salud y se le realiza la siguiente paraclínica:
Tomografía axial de órbitas que informa: alteracion en la densidad del humor acuoso del ojo izquierdo de mayor densidad que el contralateral (sangrado del humor acuoso). No se visualizan alteraciones del continente ni del contenido orbitario. Nervios ópticos con flexibilidad y espesor normales. Resto del examen normal. Se sugiere completar valoración con resonancia magnética orbitaria.
Resonancia magnética de cráneo con enfoque de órbitas y senos faciales que muestra: disminución de volumen del globo ocular izquierdo con hermorragia subaguda a nivel del humor vítreo asociada a pequeña imagen nodular en la retina. Se interpreta que podría tratarse de una enfermedad de Coats a controlar.
Ecografía ocular donde se ve: imagen melaniforme en cavidad vítrea póstero anterior; opacidad de cavidad vítrea inhomogénea; hemovítreo organizado y un globo ocular izquierdo con disminución de su longitud axial.
Fondo de ojo que evidencia: esquisis en el ojo izquierdo.
Electro-retinograma que muestra: disfunción bilateral intensa de la retina, que compromete sistema de conos y bastones.
La tomografía de coherencia óptica (TOC) es la técnica de referencia para el diagnóstico, en la cual se pueden ver esquisis sobre todo en la retina periférica. En nuestro paciente no contamos con este estudio.
Completada la evaluación paraclínica se otorga el alta con el diagnóstico de Retinosquisis; dados los antecedentes familiares antes mencionados se solicita consulta con Genetista.
En la genealogía elaborada por el equipo de genética destaca la presencia de dos familiares de 4to grado, por línea materna, de sexo masculino, con diagnóstico de retinosquisis (Figura 1). En ambos se realizó la secuenciación de las regiones codificantes del gen RS1 (en la Universidad de Iowa, USA, en 2014) y se informó la presencia de la variante c.466A>G (Arg156Gly).
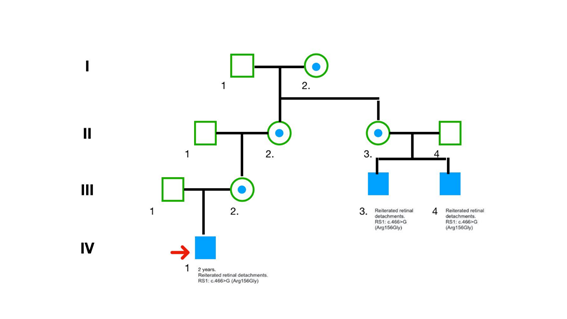
Figura 1: Genealogía donde se muestra la segregación de la mutación y su expresión fenotípica en la familia analizada, siendo compatible con el tipo de herencia ligada al cromosoma X recesiva. El paciente índice está indicado por una flecha. Los miembros de la familia afectados están marcados con símbolos llenos. Se indica con un círculo a las mujeres presumiblemente heterocigotas.
Con el planteo diagnóstico de Retinosquisis ligada al cromosoma X, se realizó la búsqueda de la variante familiar en nuestro paciente, realizando secuenciación de la región genómica de interés (ChrX:18662606, Human Genome Assembly GRCH37) a través de electroforesis capilar con secuenciador automático 3500XL y análisis de los electroferogramas con el Software Sequencing Analysis de Applied Biosistems, con lo cual se logra demostrar que presenta dicha variante en hemicigosis.
La mutación detectada c.466>G(Arg156Gly), es una transición que produce el cambio de un aminoácido básico (Arginina) a uno neutro (Glicina) en la posición 156 de la proteína.
Esta variante no está descrita en la base de datos genomAD (https://gnomos.broadinstitute.org/gene/ENSG00000102104) por lo que su frecuencia poblacional sería extremadamente baja.
Sin embargo, ha sido descrita previamente como patogénica en una familia extensa. Si bien no realizaron estudios funcionales en ese trabajo, demuestran la segregación de la variante con la enfermedad, con un patrón de herencia ligado al X. (6
La base de datos PolyPhen la predice como una variante benigna, a pesar de esto, la misma está en un residuo y una región altamente conservados (Figura 2). Se ubica en el dominio discoidín de la misma (aminoácidos 85 a 191), sector altamente conservado, esencial para el funcionamiento del complejo proteico de la Retinosquina y donde se agrupan la mayoría de las mutaciones descritas7,8 (Figura B).
Ademas del dominio discoidín (unidad estructural mayor), la proteína está compuesta de dos capas adyacentes, dos anillos homo-octaméricos que interaccionan entre sí y son estabilizados por puentes disulfuro9 (Figura A). En la estructura tridimensional del complejo proteico, enfrentando a la zona de la Arg156, está el Glu75 (de carga negativa) de la cadena vecina. Se puede especular que en caso de que se forme un puente salino en el complejo, este estaría afectado por la mutación. Este mecanismo ha sido propuesto para explicar el rol patogénico de otras variantes en RS1. Asimismo se puede inferir que, como ocurre para otras mutaciones de este tipo, se produzca un mal plegamiento de la proteína y su retención en el retículo endoplásmico.10

Figura 2: Tabla comparativa donde se muestran diferentes especies y su secuencia aminoacídica en el tramo donde se ubica la mutación. Base de datos Archive. https://grch37archive.ensembl.org/Homo_sapiens/Gene/Compara_Ortholog?db=core;g=ENSG00000102104;g1=ENSPTRG00000021713;hom_id=14178934;r=X:18658030-18690229
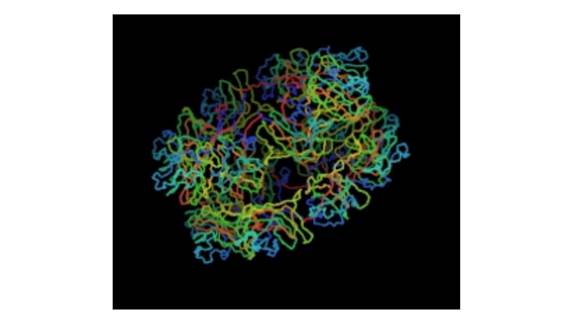
Figura 3A . Imagen tridimensional de la proteína Retinosquina donde puede verse el complejo homo-oligomérico que forma dos anillos adyacentes de ocho subunidades cada uno. El complejo se ve en una vista oblicua por lo que queda un anillo abajo a la derecha y el otro arriba a la izquierda. Gentileza del Dr. Martín Graña. Unidad de Bioinformática, Institut Pasteur Montevideo.
Figura 3B Modelo de subunidad de la proteína (una de las ocho correspondientes a cada anillo) donde se muestran algunas mutaciones conocidas, tanto en la región estructural como en el dominio discoidín. Se indica la posición de la mutación hallada en este caso (R156G) la que se ubica en la hoja plegada Beta número 4. Tomado de X-linked Juvenile retinoschisis. Molday et al.
Figura 3C Diagrama lineal de la proteína Retinosquina donde se muestran los dominios de la proteína y donde se ubican las diferentes mutaciones conocidas y la que porta nuestro paciente (R156G). Modificado de X-linked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Molday et al. (2012) y Yi et al. (2012).
Este resultado nos permitió acercar al diagnóstico genético de Retinosquisis ligada al cromosoma X, y nos habilitó a realizar asesoramiento genético a la familia, tanto para los parentales de nuestro paciente como para sus otros familiares.
Al momento, nuestro paciente ha presentado esquisis en ambos ojos, con compromiso macular solo en ojo izquierdo, por lo que presenta visión conservada en ojo derecho y cuenta dedos en ojo izquierdo.
Dado que no contamos con un tratamiento especifico para esta patología, se continúa con controles oftalmológicos reglados con el fin de detectar complicaciones (desprendimiento de retina importante) que requieran cirugía .Figura C
Discusión
La Retinosquisis es a una distrofia vítreo-retiniana caracterizada por la separación o desdoblamiento de las capas de la retina y la formación de quistes intra-retinianos.
La edad de presentación y las alteraciones retinianas características, orientan a una forma congénita de retinosquisis en este paciente. El antecedente familiar de retinosquisis causada por la mutación en el gen RS1, y la genealogía compatible con herencia ligada al cromosoma X, sugiere que presenta la misma enfermedad que sus familiares. Por esta razón se solicita la variante familiar en el paciente, se detecta y se confirma el diagnóstico planteado.
Nuestro paciente presenta una variante de baja frecuencia poblacional, en una región altamente conservada, que probablemente provoca un cambio estructural y funcional en la Retinosquina. Describimos la segunda familia en la que se muestra la segregación de la variante con el fenotipo ocular, por lo que interpretamos que es una variante patogénica, causante de la enfermedad en esta familia.
En cuanto a la transmisión de la enfermedad, asumimos que la madre del paciente es portadora de la mutación. Por lo tanto, el riesgo de tener hijos varones afectados es del 50%. Para los descendientes del paciente, se espera que sus hijas sean todas portadoras y los hijos varones sanos. Contar con el diagnóstico molecular posibilita la prevención de la transmisión de la enfermedad en esta familia mediante diagnóstico preconcepcional o prenatal, asi como la detección de mujeres portadoras de la familia.
Si bien no se plantean en este caso, los diagnósticos diferenciales a tener en cuenta son: el Síndrome de Norris ligado al Cromosoma X (presenta desprendimiento de retina al nacimiento), el Síndrome de Goldman-Favre (puede presentar retinosquisis a nivel de fóvea), la retinopatía del prematuro y la retinopatía traumática.
Las complicaciones más comunes en la evolución de la Retinosquisis son el desprendimiento de retina y las hemorragias en el vítreo. Como mencionamos anteriormente, nuestro paciente presentó esquisis a nivel macular en ojo izquierdo y periférica en ojo derecho, por lo que actualmente presenta visión cuenta dedos a izquierda y visión conservada a derecha.
Hasta el momento el tratamiento se basa en el control oftalmológico exhaustivo con el fin de detectar complicaciones pasibles de sanción quirúrgica, como medida paliativa, no existiendo un tratamiento curativo para esta afección. Sin embargo un trabajo presentado por Apushkin et al en 2006 muestra una importante mejoría de la visión con la aplicación tópica de Dorzolamide 2% en pacientes con afectación a nivel de fóvea. (11 Además, grupos como los de Janssen et al. (2008) y Park et al. (2009), han presentado trabajos de terapia génica en ratones knockout utilizando Adenovirus como vector, a través de la injección sub-retiniana e intra-vítreo respectivamente, cuyos resultados son prometedores. (12,13
En resumen, la Retinosquisis ligada al cromosoma X es una patología poco frecuente, pero de gran importancia por su impacto en la calidad de vida. En este caso se confirmó el diagnóstico a nivel molecular lo que permite un adecuado asesoramiento genético y realizar estrategias de prevención de la transmisión de esta mutación. Además, dado que se reporta el caso de la segunda familia en la que se demuestra la segregación de esta variante con la enfermedad, aportamos un fuerte argumento a favor de su rol patogénico. Conocer las diferentes mutaciones y su efecto fenotípico ayuda a entender mejor la función de esta proteína y cómo se genera la patología, y permite investigar posibles tratamientos dirigidos.

