










Serviços Personalizados
Journal

Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkOdontoestomatología
versão impressa ISSN 0797-0374versão On-line ISSN 1688-9339
Odontoestomatología vol.23 no.38 Montevideo 2021 Epub 01-Dez-2021
https://doi.org/10.22592/ode2021n37e306
Articles
Molecular mechanisms of amelogenesis imperfecta. A review of the ENAM, AMBN, FAM83H, MMP20, and KLK4 genes
1
 http://orcid.org/0000-0002-6791-8911
http://orcid.org/0000-0002-6791-8911
2
http://orcid.org/0000-0003-1872-9644
1Facultad de Odontología, Universidad de la República, Uruguay.
2Área de Patología Molecular Estomatológica, Facultad de Odontología, Universidad de la República, Uruguay. vanesapereira91@hotmail.com
Amelogenesis imperfecta (AI) is an inherited disorder that affects the structure and clinical appearance of tooth enamel. To date, mutations of 18 genes have been associated as the etiology of non-syndromic AI. This study aims to update the current knowledge on the ENAM, AMBN, FAM83H, MMP20, and KLK4 genes that cause the different types of AI.
Methodology:
The literature review included scientific articles from 2003 to 2021 on specific mutations in the genes mentioned above. SciELO, Pubmed/MEDLINE, Cochrane, and Springer Link were the databases selected.
Conclusions:
Enamel alterations can have a variety of characteristics depending on the gene involved. The biological mechanisms that lead to the disease are multiple and varied; however, many of them are not entirely clear yet, so more research is necessary to improve our understanding of the subject.
Keywords: amelogenesis imperfecta; hypoplasia; enamel
La amelogénesis imperfecta (AI) es un trastorno hereditario que afecta la estructura y apariencia clínica del esmalte dental. Hasta la fecha, se han asociado las mutaciones de 18 genes como la etiología de la AI no sindrómica. El objetivo de este trabajo es actualizar los conocimientos vigentes acerca de los genes ENAM, AMBN, FAM83H, MMP20 y KLK4 causantes de los diferentes tipos de AI.
Metodología:
Se realizó una búsqueda bibliográfica considerando artículos científicos desde el 2003 al 2021 sobre mutaciones en los genes mencionados en los siguientes portales: scielo, Pubmed/MEDLINE, Cochrane y Springer Link.
Resultados:
37 artículos cumplieron los criterios de inclusión y fueron utilizados para esta revisión.
Conclusiones:
Dependiendo del gen implicado, las alteraciones del esmalte pueden mostrar una variedad de características. Los mecanismos biológicos que conducen a la enfermedad son múltiples y variados, sin embargo, muchos de ellos no están del todo claro aún, por lo que se requerirá de más investigaciones para mejorar nuestra comprensión del tema.
Palabras clave: amelogénesis imperfecta; hipoplasia; esmalte
A amelogênese imperfeita (AI) é uma doença hereditária que afeta a estrutura e aparência clínica do esmalte dentário. Mutações de 18 genes têm sido associadas como causa do AI. O objetivo deste trabalho é atualizar o conhecimento atual sobre genes ENAM, AMBN, FAM83H, MMP20 e KLK4 que causam os diferentes tipos de IA.
Metodologia:
Foi realizada uma busca bibliográfica considerando artigos científicos de 2003 até 2021 sobre mutações específicas nos genes citados nos seguintes portais: scielo, Pubmed / MEDLINE, Cochrane e Springer Link.
Resultados:
37 artigos atenderam aos critérios de inclusão e foram utilizados para o desenvolvimento desta revisão.
Conclusões:
Dependendo do gene envolvido, as alterações do esmalte podem apresentar uma variedade de características. Os mecanismos biológicos que levam à doença são múltiplos e variados, porém muitos de les ainda não estão totalmente esclarecidos, portanto, mais pesquisas serão necessárias para melhorar nossa compreensão do assunto.
Palavras-chave: amelogênese imperfeita; hipoplasia; esmalte
Introduction
Alterations may occur during tooth development, specifically during amelogenesis, which will later become dental enamel defects. Amelogenesis imperfecta (AI) is one of these alterations. It is defined as a heterogeneous group of hereditary defects in the functioning of ameloblasts and the mineralization of the enamel matrix which produces teeth with multiple defects in this layer, either generalized or localized, and can affect both primary and permanent dentition.1
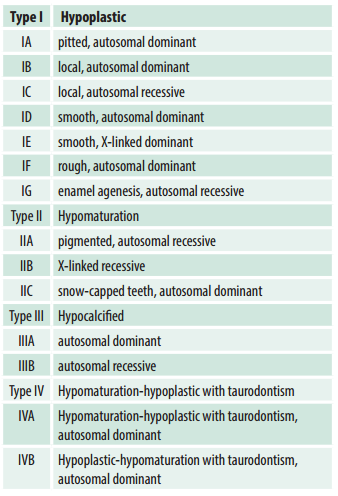
Although many classifications have been proposed for AI, the most widely used at present is Witkop's.2 He divides AI into four main groups: hypoplastic, hypocalcified, hypomaturation, and hypomaturation-hypoplastic with taurodontism, depending on the clinical presentation of the enamel defects. Each major AI group can be further divided into several subgroups according to the mode of inheritance: autosomal dominant, autosomal recessive, or X-linked2,3 (Table 1). The parameters used for this classification are thickness, hardness, and texture of the affected enamel, and radiographic presentation. Several authors state that differences in these parameters help determine when the alteration occurred.4,5
In turn, the type of AI also varies according to the gene affected and the mutation present.6 The first cases were found in the genes encoding the main proteins of the enamel matrix: AMELX (amelogenin), associated with hypoplastic AI as hypocalcified, AMBN (ameloblastin), and ENAM (enamelin), associated with hypoplastic AI.7 Subsequently it was associated with more genes such as those encoding enamel proteinases, mainly MMP20 and KLK4, resulting in hypomineralized enamel.8
Currently, a large number of genes involved in various aspects of the amelogenesis process are related to non-syndromic AI: a total of 18 affected genes.6,9FAM83H is one of the most relevant, as its mutations cause severe autosomal dominant hypocalcified AI, which is believed to be the most common in North America and is the focus of multiple studies.10,11
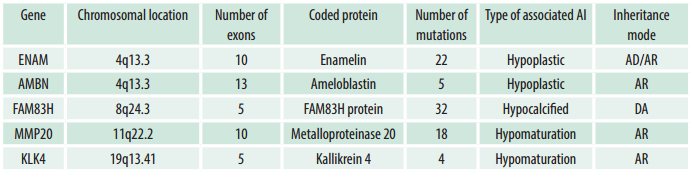
This pathology presents a complex etiology. Therefore, this study aims to update the literature on the genes most closely related to the different types of amelogenesis imperfecta: ENAM and AMBN for hypoplastic AI, FAM83H for hypocalcified AI, and MMP20 and KLK4 for hypomaturation AI.
Methodology
A search for original scientific articles was conducted in the following portals: Pubmed/MEDLINE, Cochrane, Springer Link, and SciELO. Keyword combinations were used: "amelogenesis imperfecta" plus AND operator followed by "ENAM," "AMBN," "FAM83H," "MMP20," and "KLK4". The search filters selected were Spanish and English and articles published between 2003 and 2021. We excluded articles where the genes were related to other pathologies or where AI was related to other genes. Table 2 summarizes the search strategy and its results.
Results
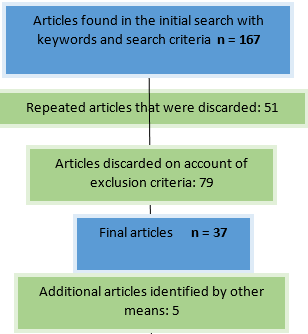
The titles and abstracts of all the articles including the key words were reviewed, and those meeting the inclusion criteria were selected for this review.
The initial search yielded 206 publications, of which 37 were included. Five additional articles identified by other means were included (references made by the authors themselves). The information collected was systematized into three sections: ENAM and AMBN causing hypoplastic AI; FAM83H causing hypocalcified AI; MMP20 and KLK4 causing hypomaturation AI. Table 3 summarizes the genes reviewed.
Hypoplastic amelogenesis imperfecta-related genes
ENAM
So far, 22 mutations in the ENAM gene detected in humans cause AI, including small deletions and insertions that shift the reading frame and truncate the encoded protein.12 The hypoplastic enamel phenotype is very varied: from small pits to horizontal grooves, mainly on the buccal faces of anterior teeth, to cases where the loss of enamel is almost complete.12,13 On radiographic examination, it is common to find a more or less thin enamel with a normal or dentin-like radiodensity, or cases where it may be absent.12,13
A normal enamel phenotype can be observed in individuals with ENAM mutations, even when other family members with the same mutation did have severe hypoplastic AI. Only ENAM mutations have presented this lack of penetrance among AI-causing mutations.13-15 Some explanations have been offered to determine this clinical variability. Recessive mutations would manifest with an additive effect, i.e., the phenotype is milder or imperceptible when only one allele is defective, compared to cases in which both are defective.14,16 Koruyucu et al. explain this variability through the location of the mutation in the terminal exon, which would result in a truncated protein with partial enamelin function. This mutation could be less harmful.13
Various alterations in the enamel architecture have been detected histopathologically. The superficial enamel can look normal, with well orientated and packed crystals, but with a general lack of structure and fusion of crystals near the dentinoenamel junction. Conversely, a normal inner layer and a structurally abnormal outer layer can be observed.17,18 Enamelin plays a significant role in the enamel's structural organization, but enamelin alterations can affect the enamel in different ways.
Brookes et al. explained this transition from normal to abnormal internal structure as driven by endoplasmic reticulum (ER) stress.18 When truncated proteins accumulate in the lumen of the ER, they trigger an unfolded protein response in an attempt to relieve stress and restore homeostasis. This disrupts ameloblast function at a given point in the secretory stage, changing normal enamel production to structurally abnormal enamel. Electron microscopy showed that the affected ameloblasts contained large intracellular vesicles with retention of enamel proteins. Homozygous mice were more severely affected as both copies of ENAM were mutated, doubling the concentration of mutated ENAM compared to heterozygotes. This could increase the ER stress generated during intracellular trafficking leading to a more severe response and cell apoptosis.18 This supports the idea that ENAM-linked AI has a dosedependent effect.14,16
AMBN
AMBN is responsible for encoding ameloblastin, one of the three main proteins of the enamel matrix, so it has always been thought that an alteration in its structure could lead to AI.19 Poulter et al. believe that the participation of ameloblastin in ameloblast adhesion would play a significant role.20 This is supported by research with mutant AMBN mice, where the ameloblast layer is detached from the enamel matrix, and the Tomes process does not develop, resulting in a hypoplastic AI phenotype.21
The scarcity of cases of AMBN mutations responsible for AI could be due to its recessive mode of inheritance.19 Another reason could be that the AMELX gene would partially compensate for the AMBN deficiency since the two genes are phylogenetically related and share some similarities.21 Therefore, compensation of ameloblastin by amelogenin could contribute to eliminating possible dental defects or make them harder to detect in homozygous mutations.19
The clinical characteristics of patients with these alterations follow a common pattern with small variations, both in temporary and permanent dentition: rough enamel of reduced thickness and susceptible to wear.20,23,24
Lu et al. examined eleven affected individuals and observed that the enamel was detached due to alterations in the dentinoenamel junction. This exposed the hypomineralized dentin, which was rapidly lost to wear. The outline of the dentinoenamel junction increased in width, and the structure of sigmoidal transitions became a straight line in appearance.24 Despite being young, some individuals also had severe periodontal problems, bone resorption, and high tooth mobility.24
Electron microscopy showed that the most severe cases had reduced enamel thickness, reduced mineral density, and absence of any normal architecture.20 In other cases, the prismatic structure of the affected parts remains practically intact, but the number of prisms decreases.20,24
AMBN null mice have the unique feature of accumulating organic material against the dentin surface, consisting mainly of amelogenin, without enamel crystal formation. Without crystal elongation, ameloblasts cannot initiate appositional growth and expand the enamel in thickness, so amelogenin accumulates against mineralized dentin after its secretion. Although amelogenin is a critical matrix constituent, it can only function properly in the context of a mineralization front that begins with the formation of the first enamel crystals, which requires ameloblastin.23
Hypomineralized amelogenesis imperfecta-related genes
FAM83H
FAM83H mutations cause autosomal dominant hypocalcified AI.11 So far, 32 FAM83H mutations have been reported, all in the region of exon 5.25 Most are missense or frameshift mutations, which lead to premature termination of translation and generate a truncated protein that includes only the N-terminal end.7,26
Several studies have determined that a change in the expressed amount of FAM83H protein does not lead to a disease phenotype, so haploinsufficiency would not cause enamel defects. In contrast, a negative or gain-of-function effect would be how the truncated protein would trigger the disease.27-29
The clinical manifestations follow a set of common patterns: a rough, soft, yellowish-brown enamel, which is susceptible to wear, causing a rapid loss of thickness, commonly preserved in small areas on the cervical and cusp tips, which can be considered a typical feature of the FAM83H mutation.25,27,29. It was also associated with dental hypersensitivity to thermal stimuli, generalized gingivitis, and increased crown fractures.26,30
Radiographically, the most remarkable feature is the similar radiodensity of enamel and dentin, and the presence of enamel of apparent normal thickness in unerupted teeth, which would indicate that the amount of protein secreted by the ameloblast is not directly influenced by FAM83H mutations.25,26,29,30
Histopathological findings in the enamel prisms include the following defects: irregular direction, a decrease in their number, and increased interprismatic gaps, which are more evident at the dentinoenamel junction.29,31 Greater retention of proteins and reduction of minerals was also detected, causing the enamel to wear easily and function as a poor thermal insulator, causing hypersensitivity.26,32
The findings related to the protein expressed by FAM83H showed that it interacts with casein kinase 1 (CK-1), located in keratin filaments in ameloblasts.27,33 It has been suggested that the FAM83H protein might regulate the organization of the ameloblast cytoskeleton as a scaffold protein and guiding CK-1 to its physiological location. FAM83H mutations would cause disorganization of the cytoskeleton by failing to recruit CK-1 to keratin filaments, a defect that would extend to desmosomes, consequently interfering with ameloblast formation and function.30,33 In turn, in vitro studies on human embryonic kidney cells and mouse dental cells detected a high concentration of the mutated FAM83H protein in the nucleus. This reduces its amount in the cytoplasm to a level below what is necessary for normal function. This nuclear localization could contribute to the incorrect organization of the keratin cytoskeleton.29-31
Hypomaturation amelogenesis imperfecta-related genes
MMP20
The MMP20 gene encodes matrix metalloproteinase 20, an enzyme that cleaves matrix proteins, mostly amelogenin, as well as enamelin and ameloblastin.34 To date, a total of 18 MMP20 mutations causing AI have been reported, which are evenly distributed throughout the 10 exons that make up the gene, except for exons 8 and 9. These are primarily missense mutations, frameshifts, or splice site mutations, most of which would degrade mRNA after transcription, resulting in a null mutated allele.35
The clinical characteristics correspond to hypomaturation AI with yellowish-brown, sometimes porous enamel, more opaque than normal, and with relative wear on the incisal edges and cusp tips.34-36 In the most severe cases, the enamel is rapidly worn away by occlusal forces after tooth eruption, leaving exposed dentin and causing tooth sensitivity.34,36
Radiographs always show an enamel pattern with slight or barely visible contrast compared to dentin marking the mineral load reduction.34-37 Enamel nanohardness testing shows that the most superficial layers, and those closest to the cementoenamel junction, have approximately 50% of the hardness of normal enamel, while the middle layer between these two is the most affected, exceeding only 10% of the normal range.35
Seymen et al. propose that the severity of the phenotype depends on whether the MMP20 mutation encodes a metalloproteinase with an active, albeit reduced, functional capacity or whether there is a complete loss of function.36 This was observed in in vitro studies where the expression of the mutated protein was the same as normal. Still, its concentration in the conditioned medium was low or non-existent, possibly due to a change in its structure that did not allow its secretion, or because it was rapidly degraded due to its instability. These cases included the most severe enamel alterations compared to mutations where the protein was found in the medium and still had partial proteolytic function.36-38 This would demonstrate that a loss of or decrease in function causes the disease.
MMP20 and KLK4 proteins have synergistic functions, cleaving enamel proteins and facilitating their removal. MMP20 begins at the secretory stage of amelogenesis, and KLK4 joins at the enamel maturation stage.4,39 In studies with single heterozygous mice for both MMP20 and KLK4 mutations, the enamel had a normal clinical appearance. However, this changed in double heterozygous mice, where enamel was altered, and surface porosity and susceptibility to wear increased. This suggests that MMP20 and KLK4 have complementary functions, and the combined alteration of both genes would cause AI.39
KLK4
KLK4 mutations result in hypomaturation type AI.40 So far, only four different mutations of this gene have been reported, making it a relatively rare cause of AI. Of these mutations, the first two are a frameshift and a missense mutation. In both of them, mRNA would be degraded after transcription.41,42 The other two are frameshifts that affect codons in the final exon and would escape the degradation system, resulting in a mutated protein.36,40 However, Seymen et al. observed that the expression of the mutated protein was greatly reduced after secretion compared to controls, making it more prone to degradation.36
The clinical presentation is typical for hypomaturation AI: teeth with normal enamel thickness but with generalized demineralization, and a slight yellowish-brown discoloration.36,40
Radiographically, the enamel's radiodensity is lower than in a normal tooth.36
Various electron microscopy and computed tomography techniques showed a difference in the structure of the inner layers of the enamel where hypomineralization is greater, especially in the dentinoenamel junction, compared to the outermost layers. Microhardness testing showed that the surface enamel is harder than the inner enamel layer.40 These results are comparable to studies in mice with null KLK4 expression, where the enamel is hypomineralized, mainly in the innermost layers, even though it is of normal thickness and the prism pattern also appears normal under the microscope.39
The compromised activity of mutated KLK4 would considerably reduce proteolysis and hinder the diffusion of residual peptides from the deeper enamel layers to the superficial ones, where they are eliminated through ameloblast endocytosis. This causes protein retention in the deep enamel, inhibiting crystal growth in the maturation stage.40
Conclusions
Mutations in the ENAM, AMBN, FAM83H, MMP20, and KLK4 genes are responsible for the different types of AI. So far, FAM83H has the highest number of reported mutations.
The clinical features of the affected enamel and the type of AI depend on the gene involved and the relevant mutation. If the clinical and radiographic appearance of the affected enamel can be correlated with the potential genetic origin of AI, it will be possible to implement more accurate diagnostic methods and make a more precise prognosis. This will also allow professionals to provide risk information to other family members.
The biological processes and mechanisms leading to AI are many and varied. Further research with a molecular approach will improve our understanding of these mechanisms and identify new mutations or genes. This new knowledge will result in improved patient care and the development of new therapies to minimize or eliminate the problems associated with AI.
REFERENCES
1. Sapp J.P; Eversole L.R; Wysocki G.P. Patología oral y maxilofacial contemporánea, 2º edición. Madrid: Elsevier, 2005: 14-16. [ Links ]
2. Witkop CJ. Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J. Oral. Pathol. 1988; 17(9-10): 547-553. [ Links ]
3. Crawford P.J; Aldred M; Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J. Rare. Dis. 2007; 2: 17. [ Links ]
4. Hu J.C; Chun Y.H; Al Hazzazzi T; Simmer J.P. Enamel formation and amelogenesis imperfecta. Cells Tissues Ogans. 2007; 186(1): 78-85. [ Links ]
5. Aldred M.J; Crawford P.J.M; Savarirayan R. Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral. Dis. 2003; 9(1): 19-23. [ Links ]
6. Prasad M.K; Laouina S; El Alloussi M; Dollfus H; Bloch-Zupan A. Amelogenesis imperfecta: 1 family, 2 phenotypes, and 2 mutated genes. J. Dent. Res. 2016; 95(13): 1457-1463 [ Links ]
7. Smith C.E.L; Poulter J.A; Antanaviciute A; Kirkham J; Brookes S.J; Inglehearn C.F; Mighell A.J. Amelogenesis Imperfecta; Genes, Proteins, and Pathways. Front. Physiol. 2017; 8: 435 [ Links ]
8. Kim J.W, Simmer JP, Lin BPL, Seymen F, Bartlett JD, Hu JCC. Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur. J. Oral. Sci. 2006; 114(suppl1): 3-12 [ Links ]
9. Simancas-Escorcia V, Natera A, Acosta de Camargo MG. Genes involved in amelogenesis imperfecta. Part I. Rev. Fac. Odontol. Univ. Antioq. 2018; 30(1): 105-120. [ Links ]
10. Lee SK, Lee KE, Jeong TS, Hwang YH, Kim S, Hu JCC, Simmer JP, Kim JW. FAM83H mutations cause ADHCAI and alter intracellular protein localization. J. Dent. Res. 2011; 90(3): 377-381 [ Links ]
11. Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am. J. Hum. Genet. 2008; 82(2): 489-494 [ Links ]
12. Zhang H, Hu Y, Seymen F, Koruyucu M, Kasimoglu Y, Wang SK, Wright JT, Havel M.W, Zhang C, Kim JW, Simmer JP, Hu JCC. ENAM mutations and Digenic Inheritance. Mol. Genet. Genomic Med. 2019; 7(10): e928 [ Links ]
13. Koruyucu M, Kang J, Kim YJ, Seymen F, Kasimoglu Y, Lee ZH, Shin TJ, Hyun HK, Kim YJ, Lee SH, Hu JCC, Simmer JP, Kim JW. Hypoplastic AI with highly variable expressivity caused by ENAM mutations. J. Dent. Res. 2018; 97(9): 1064-1069 [ Links ]
14. Seymen F; Lee K.E; Koruyucu M; Gencay K; Bayram M; Tuna E.B; Lee Z.H; Kim J.W. ENAM mutations with Incomplete Penetrance. J. Dent. Res. 2014; 93(10): 988-992. [ Links ]
15. Wang X; Zhao Y; Yang Y; Qin M. Novel ENAM and LAMB3 mutations in Chinese families with hypoplastic amelogenesis imperfecta. Plos One. 2015; 10(3): e0116514. [ Links ]
16. Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, Uygur C, Ozdemir D, Firatli S, Aren G, Firatli E. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J. Med. Genet. 2003; 40(12): 900-906 [ Links ]
17. Siddiqui S, Al-Jawad M. Enamelin directs crystallite organization at the enamel-dentine junction. J. Dent. Res. 2016; 95(5): 580-587 [ Links ]
18. Brookes SJ, Barron MJ, Smith CEL, Poulter JA, Mighell AJ, Inglehearn CF, Brown CJ, Rodd H, Kirkham J, Dixon MJ. Amelogenesis imperfecta caused by N-Terminal enamelin point mutations in mice and men is driven by endoplasmic reticulum stress. Hum. Mol. Genet. 2017; 26(10): 1863-1876. [ Links ]
19. Delsuc F; Gasse B; Sire J.Y. Evolutionary analysis of selective constraints identifies ameloblastin (AMBN) as a potential candidate for amelogenesis imperfecta. BMC Evolutionary Biology. 2015; 15: 148. [ Links ]
20. Poulter JA, Murillo G, Brookes SJ, Smith CEL, Parry DA, Silva S, Kirkham J, Inglehearn CF, Mighell AJ. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 2014; 23(20): 5317-5324 [ Links ]
21. Fukumoto S; Kiba T; Hall B; Iehara N; Nakamura T; Longenecker G; Krebsbach P.H; Nanci A; Kulkarni A.B; Yamada Y. Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J. Cell Biol. 2004; 167(5): 973-983. [ Links ]
23 Liang T; Hu Y; Smith C.E; Richardson A.S; Zhang H; Yang J; Lin B; Wang S.K; Kim J.W; Chun Y.H; Simmer J.P; Hu J.C.CAMBN mutations causing hypoplastic amelogenesis imperfecta and Ambn knockout-NLS-laczknockin mice exhibiting failed amelogenesis and Ambn tissue-specificity. Mol. Genet. Genomic Med. 2019; 7(9): e929 [ Links ]
24. Lu T; Li M; Xu X; Xiong J; Huang C; Zhang X; Hu A; Peng L; Cai D; Zhang L; Wu B; Xiong F. Whole exome sequencing identifies an AMBN missense mutation causing severe autosomal-dominant amelogenesis imperfecta and dentin disorders. Int. J. Oral Sci. 2018; 10(3): 26. [ Links ]
25. Wang SK, Zhang H, Hu CY, Liu JF, Chadha S, Kim JW, Simmer JP, Hu JCC . FAM83H and autosomal dominant hypocalcified amelogenesis imperfecta J. Dent. Res. 2021; 100(3): 293-301 [ Links ]
26. Urzua B, Martinez C, Ortega-Pinto A, Adorno D, Morales-Bozo I, Riadi G, Jara L, Plaza A, Lefimil C, Lozano C, Reyes M. Novel missense mutation of the FAM83H gene causes retention of amelogenin and a mild clinical phenotype of hypocalcified enamel. Arch. Oral Biol. 2015; 60(9): 1356-1367 [ Links ]
27. Wang SK, Hu Y, Yang J, Smith CE, Richardson AS, Yamakoshi Y, Lee YL, Seymen F, Koruyucu M, Gencay K, Lee M, Choi M, Kim JW, Hu JCC, Simmer JP. Fam83h null mice support a neomorphic mechanism for human ADHCAI. Mol. Genet. Genomic Med. 2015; 4(1): 46-67 [ Links ]
28. Wang S.K; Hu Y; Smith C.E; Yang J; Zeng C; Kim J.W; Hu J.C.C; Simmer J.P. The enamel phenotype in homozygous Fam83h truncation mice. Mol. Genet. Genomic Med. 2019; 7(6): e724. [ Links ]
29. Zheng, Y; Lu T; Chen J; Li M; Xiong J; He F; Gan Z; Guo Y; Zhang L; Xiong F. The gain-of-function FAM83H mutation caused hypocalcification amelogenesis imperfecta in a chinese family. Clin. Oral Invest. 2021; 25(5): 2915-2923 [ Links ]
30. Xin W; Wenjun W; Man Q; Yuming Z. Novel FAM83H mutations in patients with amelogenesis imperfecta. Sci. Rep. 2017; 7(1): 6075. [ Links ]
31. Yu S; Quan J; Wang X; Sun X; Zhang X; Liu Y; Zhang C; Zheng S. A novel FAM83H mutation in one Chinese family with autosomal-dominant hypocalcification amelogenesis imperfecta. Mutagenesis. 2018; 33(4): 333-340. [ Links ]
32. Zhang C; Song Y; Bian Z; Ultrastructural analysis of the teeth affected with amelogenesis imperfecta resulting from FAM83H mutations and review of the literatures. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2015; 119(2): e69-76. [ Links ]
33. Kuga T, Sasaki M, Mikami T, Miake Y, Adachi J, Shimizu M, Saito Y, Koura M, Takeda Y, Matsuda J, Tomonaga T, Nakayama Y. FAM83H and casein kinase I regulate the organization of the keratin cytoskeleton and formation of desmosomes. Sci. Rep. 2016; 6: 26557 [ Links ]
34. Gasse B, Prasad M, Delgado S, Huckert M, Kawczynski M, Garret-Bernardin A, Lopez-Cazaux S, Bailleul-Forestier I, Manière MC, Stoetzel C, Bloch-Zupan A, Sire JY. Evolutionary analysis predicts sensitive positions of MMP20 and validates newly-and previously-identified MMP20 mutations causing amelogenesis imperfecta. Front. Physiol. 2017; 8: 398 [ Links ]
35. Wang S.K; Zhang H; Chavez M.B; Hu Y; Seymen F; Koruyucu M; Kasimoglu Y; Colvin C.D; Kolli T.N; Tan M.H; Wang Y.L; Lu P.Y; Kim J.W; Foster B.L; Bartlett J.D; Simmer J.P; Hu J.C.C. Dental malformations associated with biallelic MMP20 mutations. Mol. Genet. Genomic Med. 2020; 8(8): e1307. [ Links ]
36. Seymen F, Park JC, Lee KE, Lee HK, Lee DS, Koruyucu M, Gencay K, Bayram M, Tuna EB, Lee ZH, Kim YJ, Kim JW. Novel MMP20 and KLK4 mutations in amelogenesis imperfecta. J. Dent. Res. 2015; 94(8): 1063-1069 [ Links ]
37. Kim YJ, Kang J, Seymen F, Koruyucu M, Zhang H, Kasimoglu Y, Bayram M, Tuna-Ince EB, Bayrak S, Tuloglu N, Hu JCC, Simmer JP, Kim JW. Alteration of exon definition causes amelogenesis imperfecta. J. Dent. Res. 2020; 99(4): 410-418 [ Links ]
38. Kim YJ, Kang J, Seymen F, Koruyucu M, Gencay K, Shin TJ, Hyun HK, Lee ZH, Hu JCC, Simmer JP, Kim JW. Analyses of MMP20 missense mutations in two families with hypomaturation amelogenesis imperfecta. Front. Physiol. 2017; 8: 229. [ Links ]
39. Hu Y, Smith CE, Richardson AS, Bartlett JD, Hu JC, Simmer JP. MMP20, KLK4, and MMP20/KLK4 double null mice define roles for matrix proteases during dental enamel formation. Mol. Genet. Genomic Med. 2015; 4(2): 178-196 [ Links ]
40. Smith CEL, Kirkham J, Day PF, Soldani F, Mcderra EJ, Poulter JA, Inglehearn CF, Mighell AJ, Brookes SJ. A fourth KLK4 mutation is associated with enamel hypomineralisation and structural abnormalities. Front. Physiol. 2017; 8: 333 [ Links ]
41. Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, Wright JT. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J. Med. Genet. 2004; 41(7): 545-549 [ Links ]
42. Wang SK, Hu Y, Simmer JP, Seymen F, Estrella NMRP, Pal S, Reid BM, Yildirim M, Bayram M, Bartlett JD, Hu JCC. Novel KLK4 and MMP20 mutations discovered by whole-exome sequencing. J. Dent. Res. 2013; 92(3): 266-271 [ Links ]
Declaration of interests: The authors certify that they have no commercial interests that represents a conflict of interests in connection with the manuscript
Authors' contribution note: 1.Conception and design of study 2.Acquisition of data 3.Data analysis 4.Discussion of results 5.Drafting of the manuscript 6.Approval of the final version of the manuscript. NP: ha contribuido en a, b, c, d, e, f. AC: ha contribuido en a, d, e, f. RBM: ha contribuido en a, e, f. VPP: ha contribuido en a, d, e, f.
Received: July 15, 2021; Accepted: August 31, 2021
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
