










Serviços Personalizados
Journal

Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Medicina Interna
versão impressa ISSN 0250-3816versão On-line ISSN 1688-423X
Arch. Med Int vol.37 no. 3 Montevideo nov. 2015
Caso Clínico de Interés
Epidermólisis ampollosa congénita: a propósito de un caso
Congenital epidermolysis bullosa: report of a case
Dr. Fernando Tabares
Residente Medicina Interna
Dra. Carolina Díaz
Residente Medicina Interna
Dra. Valentina Más
Asistente Clínica Médica
Dra. Raquel Monteghirfo
Profesora Adjunta Clínica Médica
Recibido: 08/07/15 - Aceptado: 12/10/15
Institución responsable: Clínica Médica 3 (Facultad de Medicina - UdelaR). Hospital Maciel. ASSE.
Correspondencia: Dra. Raquel Monteghirfo. Dirección: 25 de mayo 174. Montevideo, Uruguay.
Resumen: Arch Med Interna 37(3):
La Epidermólisis ampollosa es una enfermedad hereditaria, crónica, incurable y de baja prevalencia. Se caracteriza por la aparición de ampollas luego de traumatismos mínimos, de manifestación predominantemente cutánea y de difícil manejo. Interfiere en la calidad de vida del paciente requiriendo un abordaje terapéutico multi-disciplinario.
Presentamos a continuación un caso clínico, con el objetivo de describir manifestaciones clínicas, el enfoque diagnóstico y las posibilidades terapéuticas actuales para el abordaje integral de los pacientes que padecen esta enfermedad.
Palabras clave: Epidermólisis, enfermedad ampollar, enfermedad congénita.
Abstract: Arch Med Interna 37(3):
Epidermolysis bullosa is an hereditary, chronic,disease, incurable and with very low prevalence. It is characterized by blisters after minor trauma, with predominantly cutaneous manifestation and difficult manage. Interferes with the quality of life of patients requiring a multi-disciplinary therapeutic approach.
We present a clinical case, in order to describe clinical manifestations, current diagnostic approach and therapeutic possibilities for the comprehensive management of patients with this disease.
Keywords: epidermolysis, bullous disease, congenital disease.
INTRODUCCION
La Epidermólisis ampollosa es una enfermedad genética, hereditaria, crónica, sin tratamiento específico (1). Tiene una prevalencia muy baja, en Estados Unidos la misma es de 8/millón de habitantes (2) y se estima para Uruguay 1 de cada 100.000 habitantes (3). Se caracteriza por la aparición de ampollas luego de traumatismos mínimos, de manifestación predominantemente cutánea y de difícil manejo. La lesión cutánea tiene en la base de su patogenia alteraciones en las proteínas de anclaje de la epidermis con la dermis (1,2,4 ). El diagnóstico es ultraestructural, de biología molecular y genético. Los criterios de clasificación de esta enfermedad han sido revisados recientemente y se basan en el plano donde se forma la ampolla, la distribución de las lesiones y la identificación de la proteína afectada por inmunofluoresencia (5). Esta enfermedad interfiere en forma importante en la calidad de vida del paciente requiriendo un abordaje terapéutico multi-disciplinario existiendo en la actualidad varios tratamientos en estudio, con resultados prometedores.
Dada la complejidad que puede llegar a presentar estos pacientes para su manejo es que traemos a presentar este caso clínico. El objetivo de esta presentación es describir las manifestaciones clínicas, el enfoque diagnóstico y las posibilidades terapéuticas actuales para el abordaje integral de los pacientes que padecen esta enfermedad.
CASO CLINICO
SM, 18 años. Mal medio socioeconómico. Situación de calle. Antecedente personal de Epidermólisis ampollosa congénita diagnosticada desde el nacimiento en Hospital Pereira Rosell. Múltiples internaciones por empujes de su enfermedad. Adicto a pasta base.
Ingresa por un cuadro de 4 meses de evolución dado por aumento de sus lesiones de piel en número y tamaño. Comienza con flictenas con contenido serofibrinoso y serohemático, sin supuración, que evolucionan a erosiones, costras y posteriormente cicatrices atróficas, en miembros inferiores y superiores, en zonas expuestas a fricción. No presenta signos fuxivos perilesionales así como tampoco compromiso mucoso. Al examen físico se encuentra en apirexia y presenta flictenas, erosiones, ulceras y costras en dedos, palma y dorso de ambas manos, antebrazos y brazos. También en plantas, dedos y dorso de ambos pies y en ambas piernas. Las lesiones se encuentran en diferentes estadíos evolutivos (Figura 1 y 2). No presenta signos fluxivos en las zonas afectadas de piel así como tampoco supuración. Las mucosas están sanas. El examen cardiovascular y respiratorio del paciente es normal.
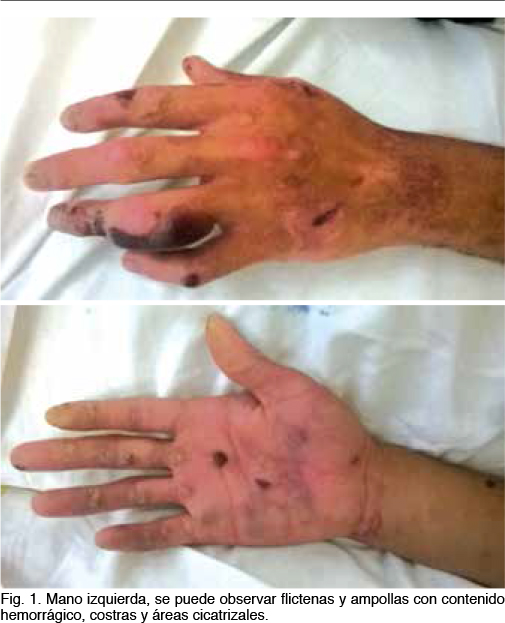
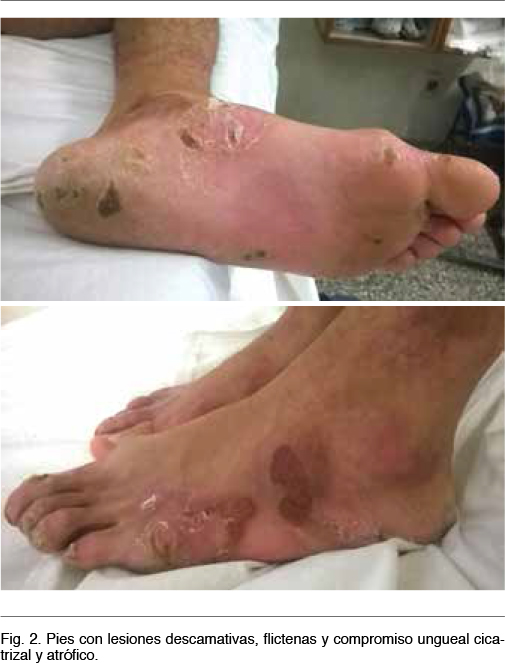
No se solicitaron exámenes de laboratorio para la confirmación diagnóstica, ya que el paciente ya tiene el mismo desde el nacimiento.
Por las características descriptas de las lesiones de piel se plantea empuje de su enfermedad genética, hereditaria, sin complicaciones infecciosas en este momento. Se trata de un tipo clínico generalizado ya que es de causa congénita y visto la imposibilidad de acceder a los estudios de microscopía electrónica realizados en el Hospital Pereira Rossell al momento del diagnóstico, no fue posible determinar el subtipo de su enfermedad según la topografía de la lesión.
El paciente fue dado de alta precozmente con medidas profilácticas para prevenir la aparición de nuevas lesiones por procedimientos inadecuados para el paciente en un centro hospitalario donde no se cuenta con personal entrenado en este tipo de patología y para evitar complicaciones infecciosas intrahospitalarias.
DISCUSION Y COMENTARIOS
Las epidermólisis ampollosas (EA) las podemos agrupar dentro de las enfermedades ampollares. Se caracterizan por una fragilidad excesiva de piel y mucosas a las fuerzas de fricción resultando en la formación de ampollas. Los sitios más afectados son aquellos expuestos a la fricción, por lo que se les ha denominado enfermedades mecáno- ampollosas (1), como pueden verse en el caso clínico presentado. Puede ocurrir frecuentemente afección extra cutánea con compromiso de dientes, tracto gastrointestinal, besico-urinario y pulmonar aunque nuestro paciente no presentaba evidencia clínica de estas afectaciones.
El mayor registro de prevalencia e incidencia de la EA, extraído del registro nacional de EA surge de un estudio longitudinal transversal de 3300 pacientes con EA en EEUU desde 1986 al 2002. La prevalencia fue estimada en 8/ millón de habitantes y la incidencia fue estimada en 19/ millón de habitantes. Se estima para Uruguay una prevalencia de 1 cada 100.000 habitantes. La relación hombre / mujer es muy similar, no existiendo predilección por sexo así como tampoco por la raza (2,3,4).
Pueden ser congénitas o adquiridas. La EA adquirida, se presenta aproximadamente en la quinta década de la vida y ocurre en forma secundaria a la producción de auto-anticuerpos contra el colágeno. La forma congénita, se engloba dentro de un grupo de enfermedades hereditarias, se presenta en la etapa neonatal y se produce por alteraciones en proteínas que intervienen en la unión de la epidermis con la dermis. En esta revisión nos referiremos a esta última, por ser esta la que presentaba nuestro paciente (1,2,4).
Clasificación
La EA se clasifica, según el nivel de la piel en donde se produce la ampolla, en tres grandes grupos: EA simple (EAS) en la cual la ampolla se localiza en la capa basal de la epidermis, EA de la unión o juntural (EAJ) donde la ampolla se localiza a nivel de la unión dermo- epidérmica y la EA distrófica (EAD) en donde la separación ocurre a nivel de la dermis (6). Figura 3
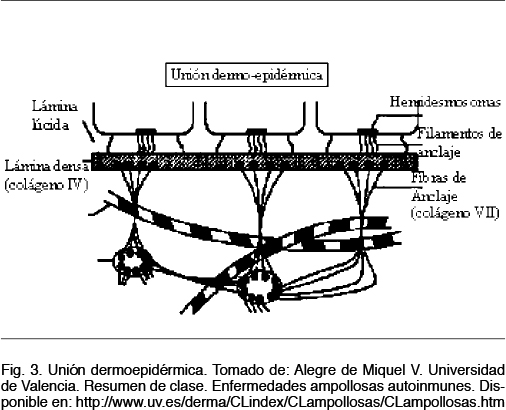
Dentro de las distintas formas de EA, la EAS es la presentación más frecuente (92 %), seguida por la EAD (5 %) y finalmente por la EAU (1 %) (1).
En 2013 se revisaron los criterios de clasificación de EA, por lo que existen distintos sistemas para la misma (7). El primero, se basa en el lugar de formación de la ampolla, dividiéndolas en EAS, EAJ, EAD y el Síndrome de Kindler. Este se caracteriza por la formación de ampollas en múltiples planos (intra epidérmica, lámina lúcida y lámina densa). No contamos con el informe de microscopía electrónica del momento en que se realizó el diagnóstico al paciente y no se le realizó nueva biopsia por tener el paciente la enfermedad ya diagnosticada.
La segunda clasificación toma en cuenta la distribución de las lesiones (localizada o generalizada), la severidad y presencia o ausencia de compromiso extra cutáneo.
La última clasificación se basa en la identificación de la proteína afectada mediante inmunofluorescencia (5).
Patogenia
Para poder entender la patogénesis es necesario hacer un breve repaso de las estructuras básicas de la piel. La epidermis, se compone de múltiples capas, desde una basal de queratinocitos hasta una capa superficial de células córneas. La dermis, se compone de células dispersas en una abundante matriz extracelular (8). La unión dermo epidérmica, consta de una membrana basal que cumple con la función de mantener la adhesión entre estas (9).
En cuanto a la epidermis, el citoesqueleto de los queratinocitos basales está constituido principalmente por queratina 5 y queratina 14. Las células epidérmicas se encuentran unidas por los desmosomas y puentes de unión intercelulares. La membrana basal epidérmica se divide en tres componentes: los hemidesmosomas, la lámina lúcida y la lámina densa. Los hemidesmosomas constituyen la unidad principal de adhesión en la unión dermo-epidérmica de la piel. Los filamentos de queratina se insertan en los hemidesmosomas mediante proteínas de la familia de las plaquinas. Las plaquinas se asocian a su vez con dos proteínas transmembranales contenidas en la placa externa de los hemidesmosomas: la a6b4 integrina y el antígeno penfigoideampolloso de 180 kDa (BP180 o BPAG2), también conocido como colágeno XVII (constituye un filamento de anclaje). La a6b4 integrina presenta dos dominios uno que se extiende hasta la lámina lúcida, y otro hasta la lámina densa. El principal componente de las fibrillas de anclaje es el colágeno VII. Los extremos de colágeno VII se insertan en la lámina densa, donde interactúan con los filamentos de anclaje por medio de su asociación con el colágeno IV (1).
Gracias a los estudios de biología molecular y genética se han identificado distintas mutaciones en los subtipos de EA. Dentro de éstas destacamos básicamente mutaciones en los genes de las queratinas basales 5 y 14, tal es el caso de EAS. En la EAJ, existen alteraciones en determinados componentes de los hemidesmosomas como la laminina 5, integrina a6b4, y el antígeno BP180 (o colágeno XVII); y en la EAD existen mutaciones en el colágeno tipo VII, componente mayoritario de las fibrillas de anclaje (5).
Por último, debemos recordar que la gravedad de las manifestaciones clínicas varía mucho entre los tipos y subtipos de EA, abarcando un espectro que va desde la formación de ampollas pequeñas hasta la presencia de ampollas muy extensas, que pueden en algunos casos ocasionar la muerte del paciente. No es posible adjudicar estas diferencias en la expresión clínica a un único gen mutado, e incluso una misma anormalidad genética puede asociarse con manifestaciones clínicas diferentes (1).
Presentación clínica
La EA se presenta como ampollas localizadas o generalizadas, las cuales pueden curar con o sin cicatriz, aparecen frente a traumatismos y/o fuerzas de fricción y característicamente cursan con el Signo de Nicolski positivo (las capas superiores de la piel se desprenden de las capas inferiores cuando se frotan ligeramente). Pueden presentar dolor así como síntomas extra cutáneos oculares, respiratorios, urológicos y/o gastroenterológicos. El caso clínico analizado se presentó con afectación exclusivamente cutánea, generalizada, sin compromiso de otros parénquimas.
En la EAS las ampollas aparecen al nacer, tras eventos traumáticos y al romperse no dejan atrofia ni cicatriz (1, 5). La forma más frecuente es la localizada la cual se limita a manos y pies, no afectándose el cabello y los dientes (5). De sus subtipos se destaca la EAS herpetiformis (Downing - Meara) por su mayor mortalidad asociada a sepsis (6). Las lesiones se disponen en grupo, se produce engrosamiento de uñas, quebranto dérmico palmo-plantar tras reiterados episodios, alopecia y afección laríngea frecuente (1, 5).
La EA juntural como regla general presenta ampollas con contenido hemático en el adulto, erosiones cutáneas, distrofia cutánea, hipoplasia del esmalte dental y caries, dejando al desaparecer cicatrices atróficas (5). La variante EAJ Herlitz presenta elevada mortalidad, siendo característica la afección de la mucosa oral y laríngea (estenosis laríngea), estenosis del meato urinario en 10% de los pacientes (5), retraso del crecimiento y anemia multifactorial (1,6). La EAJ no Herlitz se diferencia de la anterior por mejor pronóstico, no cursa con retraso crecimiento ni anemia tan severos (1).
La EAD presenta ampollas en piel y mucosas que curan con cicatriz. Puede ser de herencia autosómica dominante o recesiva. La primera se presenta desde el nacimiento o edades posteriores, las mucosas y uñas a menudo son afectadas (5, 6). La EAD de herencia autosómica recesiva es más grave, menos frecuente, con ampollas desde el nacimiento, áreas de ausencia congénita de piel, fusión de los dedos tras reiterados episodios ampollas y contractura en flexión de muñecas y afección de mucosa oral, faríngea, esofágica y ano. Desnutrición y retraso crecimiento por alteraciones mucosas son frecuentes (1).
El Síndrome de Kindler se manifiesta con ampollas de distribución acral, fotosensibilidad en período neonatal e infancia, poiquilodermia y telangiectasias. Se han reportado estenosis genitourinaria y gastrointestinales (5).
La complicación cutánea que determina la mayor causa de muerte en estos pacientes es el carcinoma espinocelular.
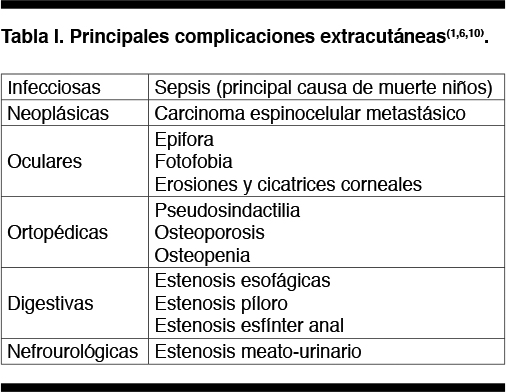
Se describen múltiples complicaciones que se enumeran en la figura 4 (1,6,10).
Diagnóstico
El diagnóstico es clínico, histopatológico, biopatológico y genético. Se realiza mediante biopsia de piel, con anticuerpos BP230, expresión de Anticuerpo anti colágeno VII como puede verse en EAS y EAD (6), así como alteraciones en queratina14 y laminina 332 (1, 11).La biopsia de la piel debe realizarse en la piel sana que rodea una ampolla reciente o friccionando piel sana para provocar una ampolla nueva. De esta manera se evitan los cambios de las proteínas secundarios a la herida o a la cicatrización (3,11). La valoración por microscopía electrónica busca determinar el nivel de separación de ampollas en relación con las capas de la piel (11,12). El mapeo antigénico en inaccesible en nuestro medio.
De sospecharse EA autoinmune puede solicitarse mapeo por inmunofluorescencia, buscando autoanticuerpos a nivel de la piel perilesional, e indirecta (Auto-anticuerpos circulantes) no siendo este el caso de nuestro paciente (13).
Tratamiento
No existe ningún tratamiento efectivo, siendo los mismos sintomáticos y paliativos. El mismo requiere un equipo multidisciplinario (dermatólogo, internista, fisioterapeuta, nutricionista, odontólogo y psicólogo) (1,3,11).
Todo paciente en el que se realice diagnóstico de EA congénita debe ser derivado en nuestro país al centro de Enfermedades Raras que se tratan en el Banco de Previsión Social (BPS). Ahí serán seguidos por un equipo multidisciplinario entrenado en el manejo de esta patología (3).
Para las lesiones cutáneas se realizan curaciones diarias, pudiendo requerir antibióticos tópicos (mupirocina, ácido fusídico o gentamicina) en caso de presentar lesiones sobreinfectadas; en cuyo caso se deberá tomar muestras de cultivo y antibiograma, debido a que se observa alta tasa de resistencia por los múltiples planes antibióticos utilizados en estos pacientes. Las ampollas pueden puncionarse para evitar crecimiento de las mismas, siempre por su base, sin retirar el techo (1, 11).
Se han visto mejores resultados en tratamientos con apósitos húmedos cuando hay ampollas que la cura tradicional, utilizándose además apósitos no adhesivos de tipo hidrocelulares, de espuma de poliuretano, de siliconas o de tul vaselinado (3).
Hay que tratar de evitar los traumatismos de la piel que provocan las punciones para toma de muestra de exámenes de laboratorio o fijación con leucoplasto de curaciones para evitar la formación iatrogénica de ampollas, a menos que sea estrictamente necesario.
El manejo del dolor es de vital importancia requiriendo tratamiento psicológico, analgésicos y sedantes, pudiendo necesitar inclusive anestesia regional (14).
En lo odontológico se recomiendan controles frecuentes, limpieza anual o bi-anuales con aplicación de flúor y restauración dental bajo anestesia (1).
El tratamiento quirúrgico está destinado a liberación de pseudosindactilia y contractura en flexión.
Se sugiere realización de interconsulta con oftalmólogo por eventual necesidad de lágrimas de metil-celulosa profilácticas y antibióticos tópicos en caso de erosiones (1,6).
Se encuentran en estudio nuevas terapias, que evidencian resultados prometedores:
- Terapia proteica: consiste en aplicación de proteína faltante, incentiva la curación más rápida de las heridas. Buenos resultados en pruebas animales.(1,15)
- Terapia celular:
1- Fibroblastos: estimulan la formación de colágeno VII a nivel de la unión (Fase I, pruebas humanos) (1, 15)
2- Células del estroma mesangial: mejora la curación de las heridas y expresión de colágeno (1,15,16 )
3- Células Hematopoyéticas: los trasplantes de células hematopoyéticas en modelos animales migran hacia la piel y se diferencian en células epidérmicas con formación de queritocitos (1,15,17)
- Terapia genética: consiste en transferencia de genes, mediante vectores virales (más efectivos) y no virales (plásmidos). Recientemente se han publicado estudios con pruebas en humanos y buenos resultados. Consiste en obtención de piel y conversión en células madre pluripotenciales con reparación del defecto genético (mutación COL7a1) y reimplantación en los pacientes (1,15,16,18).
Control y seguimiento
Estos pacientes deben ser controlados por el equipo de enfermedades raras del BPS 2 veces al año, prestando especial atención al diagnóstico precoz del carcinoma espinocelular que determina la mayor causa de muerte en esta población. Figura 4.
CONCLUSIONES
Hemos visto un paciente portador de enfermedad ampollar congénita generalizada, patología muy poco frecuente, cursando empuje de la misma, sin complicaciones infecciosas actuales A punto de partida de este caso se realizó una revisión bibliográfica incluyendo definición, nueva clasificación, patogenia, presentación clínica y abordando finalmente el tratamiento, haciendo hincapié en el manejo multidisciplinario de estos pacientes por el importante impacto que esta patología tiene sobre su calidad de vida.
BIBLIOGRAFIA
1) Siañez C, Pezoa R, Salas-Alanis J.C. Epidermólisis ampollosa congénita: revisión del tema. Actas Dermosifiliogr. 2009;100:842-56.
2) Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis 2010; 5:12.
3) Caballero G, Castro M, Fernandez L, Fuentes A, et al. Guia Clinica, tratamiento de Epidermolisis Ampollar en linea]. Montevideo: BPS; 2014 [acceso: junio 2015]. Disponible en: http://www.bps.gub.uy/bps/file/8964/1/guia_clinica_epidermolisis_bullosa.pdf
4) Fine JD, Johnson LB, Suchindran C, Carter DM, Moshell A. Epidermolysis Bullosa: Clinical, Epidemiologic, and Laboratory Advances, and the Findings of the National Epidermolysis Bullosa Registry. In: Fine JD, Bauer EA, McGuire J, Moshell A, editor. The National Epidermolysis Bullosa Registry: organization, goals, methodologic approaches, basic demography, and accomplishments. Baltimore: Johns Hopkins University Press; 1999. pp. 79–100.
5) Laimer M, Bauer J, Murrell D. Epidemiology, pathogenesis, classification, and clinical features of epidermolysis bullosa [on line]. UpToDate. Mar. 2015 [acceso junio 2015]. Disponible en : http://www.uptodate.com/contents/epidemiology-pathogenesis-classification-and-clinical-features-of-epidermolysis-bullosa
6) Baselga Torres E. Enfermedades ampollosas hereditarias. Enfermedades ampollosas [en línea]. En: Moraga Llop FA. Protocolos de Dermatología. 2da. ed. Madrid: Asociación Española de Pediatría; 2007 [acceso: junio 2015]; pp.15-22. Disponible en: https://www.aeped.es/sites/default/files/documentos/enfermedadesampollosas.pdf
7) Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad. Dermatol 2014; 70:1103-26.
8) Aumailley M, Has C, Tunggal L, Bruckner-Tuderman L. Molecular basis of inherited skin-blistering disorders, and therapeutic implications. Expert Rev Mol Med. 2006; 8: 1-21.
9) Masunaga T. Epidermal basement membrane: its molecular organization and blistering disorders. Connect Tissue Res. 2006;47: 55-66.
10) Woodley DT, Cogan J, Wang X, Hou Y, Haghighian C, Kudo G.De novo anti-type VII collagen antibodies in patients with recessive dystrophic epidermolysis bullosa.J Invest Dermatol. 2014;134:1138-40.
11) Torres MC, Contreras C, González ML. Epidermólisis ampollosa en el recién nacido - reporte de un caso. Rev CES Med. 2011; 25: 221-230.
12) Cepeda-Valdes R, Pohla-Gubob G , Borbolla-Escoboza JR, Barboza-Quintanac O, Ancer-Rodríguez J, Hintner H, et al. Mapeo por inmunofluoresencia para el diagnstico de epidermólisis ampollosa congénita. Actas Dermosifiliogr.2010; 101:673-682.
13) Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Inmunofluorescence testing in the diagnosis of autoimmune blistering diseases. An.Bras.dermatol. 2014; 89:885-9.
14) Goldschneider KR, Good J, Harrop E, Liossi C, Lynch-Jordan A, Martinez AE, et al. Pain care for patients with epidermolysis bullosa: best care practice guidelines. BMC Med. 2014;12:178.
15) Vanden Oever MJ, Tolar J. Advances in understuding and treating dystrophic epidermolysis bullosa. F1000Prime Rep. 2014; 6:35.
16) Wenzel D, Bayerl J, Nyström A, Bruckner-Tuderman L, Meixner A, Penninger JM. Genetically correcte diPSCs as cell therapy for recessive dystrophic epidermolysis bullosa. Sci Transl Med. 2014; 6:264ra165.
17) Umegaki-Arao N, Pasmooij AM, Itoh M, Cerise JE, Guo Z, Levy B, et al. Induced pluri potent stem cells from human revertant keratinocytes for thetreatment of epidermolysis bullosa. Sci Transl Med. 2014;6:264ra164.
18) Sebastiano V, Zhen HH, Haddad B, Bashkirova E, Melo SP, Wang P, et al.Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci Transl Med. 2014 Nov 26;6:264ra163.

