










Serviços Personalizados
Journal

Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Curriculum ScienTI
Curriculum ScienTILinks relacionados
Compartilhar

 Permalink
PermalinkArchivos de Medicina Interna
versão impressa ISSN 0250-3816versão On-line ISSN 1688-423X
Arch. Med Int vol.36 no. 3 Montevideo nov. 2014
ARCHIVOS DEL INSTITUTO DE NEUROLOGÍA
Análisis molecular de familias uruguayas con Enfermedad de Huntington
Molecular analysis of the Uruguayan families with Huntington’s Disease
Dr. Víctor Raggio
Profesor Agregado del Departamento de Genética, Facultad de Medicina. UdelaR. Montevideo.
Dra. Bioq. Estela Bidegain
Ayudante de Análisis Clínicos, Facultad de Química. UdelaR. Montevideo.
Mag.Quim. Marcelo Vita
Asistente de Biología Molecular, Facultad de Química. UdelaR. Montevideo.
Dr. Ricardo Buzó
Profesor Agregado de la Cátedra de Neurología, Instituto de Neurología, Facultad de Medicina. UdelaR. Montevideo.
Dr. Andrés Lescano
Posgrado de Neurología, Asistente del Departamento de Neuropsicología, Instituto de Neurología, Facultad de Medicina, UdelaR. Montevideo.
Dra. Ofrenda de Medina
Ex Profesora Adjunta de la Cátedra de Neurología, Instituto de Neurología, Facultad de Medicina. UdelaR. Montevideo.
Dr. Ignacio Amorín
Ex Asistente de la Cátedra de Neurología, Instituto de Neurología, Facultad de Medicina. UdelaR. Montevideo.
Dra. Elena Dieguez
Integrante del Grupo de Trabajo en Enfermedad de Parkinson, Instituto de Neurología, Facultad de Medicina. UdelaR. Montevideo.
Dr. Roberto Ventura
Ex Profesor Adjunto del Departamento de Neuropsicología, Instituto de Neurología, Facultad de Medicina. UdelaR. Montevideo.
Dra. María Mirta Rodríguez
Profesora del Departamento de Genética, Facultad de Medicina. UdelaR. Montevideo.
Dra. Patricia Esperón
Doctora en Química, Profesora Agregada de Biología Molecular, Facultad de Química. UdelaR. Montevideo.
Centro de Tratamiento de Parkinson y Movimientos Anormales y Departamento de Genética, Facultad de Medicina. UdelaR. Montevideo.
Correspondencia: Dr. Víctor Raggio. Sección Clínica, Departamento de Genética, Facultad de Medicina. Gral. Flores 2125,
C.P.: 11800, Montevideo, Uruguay. Tel.: 29243414 / 3469, Fax: 29249563 - vraggio@fmed.edu.uy
Recibido: 25/7/2013. Aceptado: 28/11/2013
Introducción: La Enfermedad de Huntington (EH) es un trastorno neurodegenerativo; autosómico dominante, con expresividad variable y penetrancia completa. La prevalencia estimada es entre 1-4 cada 100.000 habitantes. Es causada por expansión de tripletes CAG en el exón 1 del gen IT-15 que conduce a la síntesis de una proteína con una región de poliglutaminas expandidas que forman agregados en el núcleo celular induciendo a la apoptosis. Los alelos normales presentan un número menor a 26 tripletes CAG, y aquellos con más de 40 conducirán siempre a la enfermedad. Alelos de entre 26 a 36 repetidos se consideran normales “mutables” y de 36 a 39 repetidos generan un riesgo aumentado de desarrollar la enfermedad. Objetivo: Poner a punto el diagnóstico molecular en una población uruguaya, mediante la determinación del tamaño exacto de la mutación en personas afectadas o con sospecha clínica de EH, mediante el uso de técnicas de biología molecular. Métodos: Pacientes de la Policlínica de Enfermedad de Parkinson y Movimientos Anormales del Hospital de Clínicas. La determinación del número de repetidos se realizó mediante técnicas de amplificación del ADN por PCR y posterior análisis en geles de poliacrilamida y secuenciación. Resultados: Realizamos el diagnóstico molecular de 16 pacientes, 15 con un diagnóstico clínico previo, y uno asintomático. Se descartó el diagnóstico de EH en otros dos individuos analizados. Conclusiones: Hemos logrado la puesta a punto del estudio molecular para la enfermedad de EH por primera vez en nuestro país. Esta prueba es de gran utilidad como diagnóstico confirmatorio, etiológico o diferencial de EH.
Palabras clave: Enfermedad de Huntington, Neurogenética, Genética Clínica, Genética Molecular, Mutaciones dinámicas.
Abstract
Introduction: Huntington disease (HD) is a neurodegenerative disorder with an autosomal dominant inheritance mode, complete penetrance and variable clinical expressivity. The estimated prevalence is 1 to 4 per 100.000 individuals. It is caused by a CAG triplet expansion in exon 1 of the IT-15 gene which codes for a protein with an enlarged polyglutamine region. This leads to the formation of protein aggregates in the cell nucleus and induces apoptosis. Normal alleles show less than 26 CAG repeats, and those over 40 always lead to the disease. Alleles with 26 to 36 repeats are considered normal “mutable” alleles and those between 36 to 39, are considered in a gray zone with increased risk of developing the disease. Aims: To develop a diagnosis of HD in a uruguayan population and determine the exact size of the mutation in clinically affected subjects using molecular biology techniques. Methods: Patients were derived from Neurology Clinic of the “Hospital de Clínicas”. The determination of the CAG repeat number was done using polymerase chain reaction (PCR) technique, subsequent analysis on polyacrilamide gels and sequencing. Results: We performed the molecular diagnosis in 18 patients with clinical suspicion of HD. Fifteen of them had a previous clinical diagnosis and one had no symptoms. Besides, in two additional individuals this test allowed us to discard HD. Conclusions: A molecular diagnostic for HD disease was developed for the first time in our country. This test is of great clinical utility as a confirmatory, etiological, or differential diagnosis.
Keywords: Huntington’s disease, Neurogenetics, Clinical Genetics, Molecular genetics, Dynamic mutations.
Introducción
La enfermedad de Huntington (EH), es un trastorno neurodegenerativo progresivo del sistema nervioso central, que afecta específicamente a estructuras cerebrales como la corteza cerebral y el cuerpo estriado (caudado y putamen).
De etiología genética, presenta un patrón de herencia autosómico dominante con expresividad variable y penetrancia completa: es decir, los individuos que heredan el alelo mutado, eventualmente desarrollarán la enfermedad, a menos que mueran de otras causas antes del inicio de los síntomas. En poblaciones de origen caucásico tiene una prevalencia estimada de entre 5 y 10 afectados por cada 100.000 habitantes(1).
Su forma de presentación característica es a través de una tríada de síntomas; movimientos involuntarios (coreicos), trastornos psiquiátricos y deterioro cognitivo. Todo esto conduce a la muerte 15 a 20 años después del inicio de los síntomas. La mayoría de los pacientes inicia la enfermedad en la cuarta década de la vida, aunque en un reducido número de pacientes se presenta antes de los 20 (EH juvenil) o después de los 50 años de edad (EH de inicio tardío)(2). El período presintomático (o más correctamente, premotor) se caracteriza por cambios sutiles en la personalidad, en la cognición y en el control motor. Posteriormente aparece la fase sintomática o diagnóstica, en la que las características clínicas se hacen muy evidentes. Se manifiesta entonces la tríada de síntomas motores, cognitivos y conductuales. Son típicos los distintos trastornos del movimiento, siendo el más característico la corea, (movimientos involuntarios, fluentes e impredecibles, que afectan a los miembros, lengua, boca, o musculatura axial) y puede aparecer distonía, falta de persistencia motora, incoordinación y alteración visual. Son característicos síntomas cognitivos: la pérdida de memoria, trastornos perceptivos y espaciales. Entre los cambios conductuales se destacan cambios de personalidad y neurosis como los más frecuentes, aunque se encuentran otros como obsesión, bulimia, depresión, ideas suicidas, manías y psicosis.
La EH se debe a una mutación dinámica(3), en una región inestable del genoma, en este caso un triplete CAG, en el exón 1 del gen IT15, localizado en la región 4p16.3. El producto del gen mutado es una proteína (huntingtina) con residuos poliglutamínicos adicionales, que son causantes de agregados intranucleares en neuronas específicas. Esa expansión de trinucleótidos CAG se observa en otras enfermedades “poliglutamínicas”, que constituyen un creciente grupo de síndromes neurodegenerativos humanos(4).
Se ha establecido como alelo normal aquel que presenta un número de repetidos CAG menor a 26, y aquellos con 40 o más repetidos se consideran mutaciones patogénicas que conducirán siempre a la enfermedad. Los alelos que presentan entre 26 y 36 repetidos CAG se consideran normales “mutables” y de 36 a 39 repeticiones, en una zona gris con riesgo incrementado de desarrollar la enfermedad, pero sin una penetrancia completa. Algunos individuos sin síntomas y que muestran “un tamaño intermedio” con repeticiones de CAG entre 27 a 35 están en riesgo de transmitir la enfermedad a sus descendientes(5). Aproximadamente un 1 a 2% de la población general tiene alelos de este tamaño. La probabilidad de que la transmisión de un alelo en este rango se amplíe a un alelo con mayor número de repetidos depende de varios factores, incluyendo el tamaño del alelo, la configuración molecular de la región que rodea la repetición de CAG y el sexo del parental que lo transmite(6). El riesgo de expansión aumenta de un 6 a 10%, cuando la mutación se trasmite por vía paterna.
Una característica de estas enfermedades es el fenómeno de anticipación(7). Este fenómeno se explica por el hecho de que las repeticiones expandidas de tripletes de nucleótidos no son estables y tienden a aumentar de generación en generación. Durante la meiosis el riesgo de la expansión es más frecuente en la espermatogénesis, probablemente causado por el “patinaje” de la replicación frente a lo que ocurre en la ovogénesis(8,9). En consecuencia, para el caso de la EH, aquellos individuos que heredan de sus padres el alelo “premutado” pueden tener un repetido de CAG más largo y tienden a desarrollar síntomas a una edad más temprana(10-12). En ese sentido, un número de repeticiones de tripletes CAG de más de 60, a menudo se asocia con una aparición de la enfermedad durante la niñez o la adolescencia (EH juvenil). Sin embargo, esta correlación es menos evidente en los individuos con un rango menor de repeticiones de CAG. En estos casos el número de repeticiones sólo explica un 40-50% de la variación en la edad de inicio y el resto se ve influenciado por otros factores genéticos y ambientales(13).
Las técnicas de Biología Molecular hacen posible la detección de los portadores de la enfermedad en fases presintomáticas lo que permite una detección temprana, una estimación de riesgos reproductivos, así como el diagnóstico prenatal o preconcepcional(14).
Hemos puesto a punto el diagnóstico molecular para esta afección y lo comenzamos aplicar en pacientes con sospecha clínica de EH como diagnóstico confirmatorio, etiológico, diferencial y en algunos casos predictivos. Adicionalmente, hemos estudiado la frecuencia de repetidos CAG en la población general de nuestro medio. El principal objetivo de este trabajo ha sido determinar el tamaño exacto de la mutación en personas afectadas con la EH y a sus familiares mediante el uso de técnicas de biología molecular.
Pacientes y métodos
Pacientes
Los pacientes provinieron de la Policlínica de Enfermedad de Parkinson y Movimientos Anormales del Instituto de Neurología (Hospital de Clínicas, Facultad de Medicina, UdelaR), en su mayoría, y el resto de instituciones mutuales. Quince de estos pacientes tenían un diagnóstico clínico de EH realizado por neurólogo y genetista. Un paciente estaba asintomático, pero se consideraba en riesgo por ser familiar de primer grado de un afectado. En otros dos casos se realizó como diagnóstico diferencial para descartar la enfermedad.
Diagnóstico molecular
Se determinó el número de repetidos CAG mediante amplificación por reacción en cadena de la polimerasa (PCR) y posterior análisis de fragmentos por electroforesis.
El ADN fue extraído a partir de 3 mL de sangre periférica utilizando el kit comercial QIAamp DSP DNA Blood Mini Kit, Qiagen, siguiendo las instrucciones del fabricante.
La amplificación se llevó a cabo en un volumen de reacción de 20 µL en presencia de 0,8 mM de MgCl2, 0,8 nM de dNTPs, 0,5-1 mg de ADN, 0,2 mM de cada primer, 5% DMSO, buffer de reacción 1X y 0,75 U de enzima polimerasa Hot Start (Fermentas). Las condiciones de ciclado consistieron en una desnaturalización inicial de 5 min. a 95°C, seguido de 30 ciclos de 30 seg. a 95ºC, 30 seg. a 58°C y 30 seg. a 72°C, finalizando con una extensión final de 5 min. a 72°C.
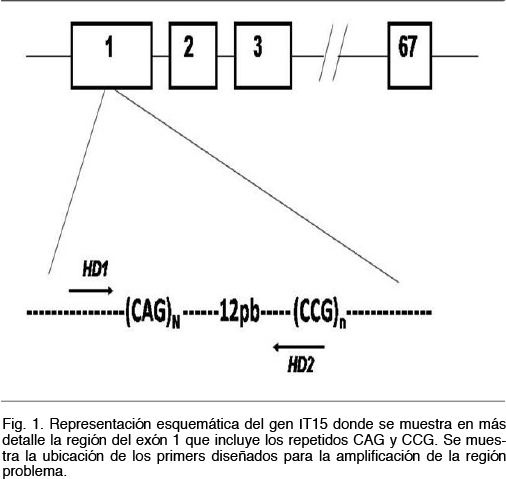
Los primeros fueron diseñados a partir de la secuencia del gen IT15 publicada en las diferentes bases de datos (Ensembl Genome Browser, www.ensembl.org). La estrategia consistió en diseñar un juego de primers que no amplificara la zona de repetidos CCG ubicada unos 12 pb en dirección 3´ de la región de los repetidos CAG. Los repetidos CCG, al igual que los CAG, son polimórficos por lo que los primers fueron diseñados para no amplificar la región que los contiene (Figura 1).
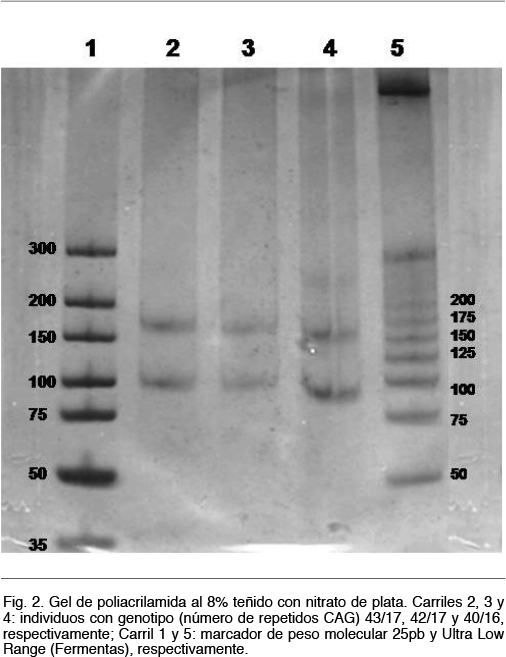
Estos fragmentos se resolvieron mediante electroforesis en gel de poliacrilamida al 8%, durante 2 horas a 80V. Se realizó la migración conjuntamente con controles que contenían un número de repetidos conocidos, un control negativo y marcadores de peso molecular. Los resultados se visualizaron mediante revelado con nitrato de plata. Los productos obtenidos serán de por ejemplo 107 pb, 128 pb y 170 pb si presentaban 19, 26 o 40 repetidos CAG, respectivamente (Figura 2). En los casos en que el número de repeticiones era cercano o superior a 40, se realizó una confirmación por secuenciación Sanger.
Resultados
Familias diagnosticadas
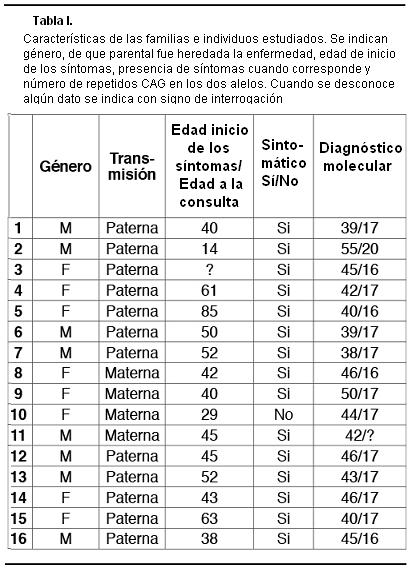
Se llegó al diagnóstico molecular de 16 pacientes pertenecientes a 8 familias diferentes, de los cuales 15 ya tenían un diagnóstico clínico, y un individuo estaba asintomático (Tabla I).
Se analizaron también dos pacientes con sospecha clínica de EH, pero en los que no se detectó un aumento del número de repeticiones CAG, por lo que se descartó la EH. En estos casos puede tratarse de fenocopias de la enfermedad de Huntington tipo 1(15) o tipo 2(16), u otras enfermedades neurogenéticas que se pueden plantear como diagnóstico diferencial de la EH.
Todos los pacientes afectados resultaron ser heterocigotas con un alelo normal y otro expandido. En los pacientes afectados de EH, el número de repeticiones CAG en los alelos mutados varió desde 40 a 55; mientras que para los alelos normales, la variación fue entre 16 y 20 repetidos CAG.
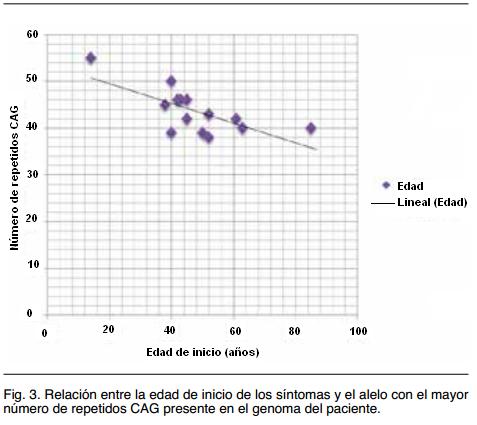
Si bien los casos estudiados son pocos y varios de ellos pertenecientes a la misma familia (por lo que comparten otros factores modificadores), se encontró una correlación inversa muy evidente entre el número de repetidos CAG y la edad de inicio de los síntomas de EH (Figura 3), como se espera de acuerdo a la fisiopatología molecular de esta afección(17).
Análisis cualitativo de una familia extensa
A modo de ejemplo se presenta esta familia (Figura 4) donde se pueden apreciar todas las características clínicas de la afección: 1. un patrón de herencia autosómico dominante con afectados en todas las generaciones y transmisión de padre a hijo varón; 2. fenómeno de anticipación, comprobado tanto a nivel clínico con una edad de comienzo de los síntomas menor y un curso con una progresividad mayor, así como a nivel molecular donde se verifica una pasaje de 39 a 55 repetidos CAG en una transmisión padre a hijo (individuos III5 y IV6); 3. una expresividad clínica y edad de comienzo variable, aún entre individuos que portan el mismo número de repetidos CAG (véase individuos: II6, III5, III9 y III10); 4. una gran variedad de expresiones clínicas, con individuos que presentan desde el inicio una corea típica y otros que comienzan con síntomas de la esfera psiquiátrica, con trastornos de personalidad y otros síntomas premotores.
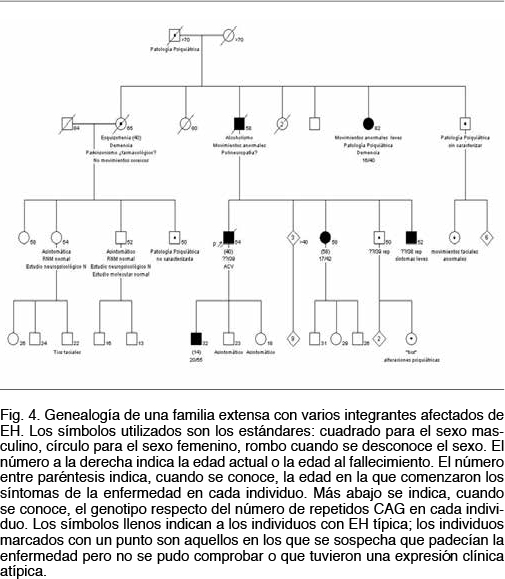
Discusión
El diagnóstico molecular puede realizarse tanto: 1. en pacientes sintomáticos, como confirmación de un diagnóstico clínico y para descartar diagnósticos diferenciales; 2. en individuos asintomáticos pero en riesgo de haber heredado la mutación, como test predictivo; 3. como diagnóstico prenatal.
El diagnóstico molecular consiste en determinar el tamaño exacto de la repetición CAG presente en el exón 1 del gen IT-15. Los alelos normales, más comúnmente reportados en la bibliografía se encuentran entre 17 y 20 repetidos, siendo la repetición de 17-19 la más frecuente en la población estudiada así como en 57 controles sanos de nuestra población. Respecto al número expandido de repeticiones la mayor frecuencia la presentan los alelos con 40 a 50 repetidos, lo que coincide con nuestros datos(18-21).
El tamaño de la expansión y la edad de inicio de los síntomas presentan una correlación ampliamente demostrada22, si bien no es el único factor determinante en la edad de inicio(23,24). En nuestra serie la tendencia a una edad de inicio más temprana cuanto mayor el número de repetidos CAG es clara (ver Figura 3).
En la mayoría de los casos en que se registra una expansión de repetidos el alelo patogénico ha sido heredado del padre. Este efecto del sexo en la transmisión de esta mutación dinámica es conocido desde hace más de 20 años(25). No contamos con estudios familiares suficientes como para comprobar esto, pero en algunos de nuestros casos este fenómeno se evidencia claramente. De hecho el único caso de EH juvenil(26) que encontramos se da por una gran expansión transmitida por vía paterna (ver Figura 4).
El problema del asesoramiento genético y la posibilidad usar test genéticos predictivos sobre la enfermedad viene siendo estudiado desde hace tiempo(27). Como se trata de una enfermedad dominante con una penetrancia de 100%, el encontrar un alelo expandido es sinónimo de que se desarrollará la enfermedad (si bien no se puede predecir con exactitud la edad de inicio y la severidad). Por otra parte en esta afección grave, de inicio habitualmente más allá de los 40 años y sin tratamiento, el tema del diagnóstico genético en individuos asintomáticos es extremadamente complejo y la realización de este tipo de estudio pronóstico en general se desaconseja(28,29). Se han desarrollado pautas sobre la aplicación y alcances de los test predictivos(30,31). De todos modos algunos individuos en alto riesgo de haber heredado la mutación (en general hijos o hermanos menores de un afectado) desean realizar el test molecular o al menos consultan con esa intención. Los motivos pueden ser por deseo de conocer los riesgos reales, para tomar decisiones de vida y, más frecuentemente, para tomar decisiones reproductivas.
En estos casos, durante una primera consulta de asesoramiento genético, se proporciona una información completa sobre la enfermedad. Se realiza una explicación del posible curso clínico de la misma, y se analizan posibilidades diagnósticas y otros aspectos relevantes para la toma de decisiones. Si una persona asintomática se somete al estudio, porque ha demostrado su voluntad expresa de conocer el diagnóstico, debe dar su consentimiento para realizarlo. El resultado de la prueba debe ser siempre entregado en el transcurso de una consulta de asesoramiento y con la colaboración de psicólogos. De ser posible, se debe involucrar a los familiares para realizar estos estudios, dado que las consecuencias pueden afectarlos.
El desarrollo de diagnósticos moleculares confirmatorios, que permiten descartar diagnósticos diferenciales y que se pueden usar de forma predictiva, preconcepcional o prenatal, son esenciales para el manejo de esta compleja y grave enfermedad, de enorme impacto en las familias afectadas y que significa uno de los mayores desafíos en Asesoramiento Genético(32).
Conclusiones
Hemos puesto a punto el diagnóstico molecular de la Enfermedad de Huntington, en el laboratorio de Biología Molecular de la Facultad de Química y lo comenzamos aplicar en pacientes con sospecha clínica de EH como diagnóstico confirmatorio, etiológico, diferencial y en algunos casos predictivo. Este diagnóstico no se realizaba en nuestro país, resultando por tanto novedoso y de utilidad para completar un diagnóstico clínico presuntivo de EH.
Bibliografía
1 Driver-Dunckley E, Caviness JN. Huntington’s disease. In Schapira AHV. Neurology and Clinical Neuroscience. Mosby Elsevier. 2007; pp. 879–85.
2 Zuccato C, Valenza M, Cattaneo E. Molecular Mechanisms and Potential Therapeutical Targets in Huntington’s Disease. Physiol Rev 2010; 90: 905-81.
3 Usdin K, Grabezyk E. DNA repeat expansions and human disease. Cell Mol Life Sci 2000; 57: 914-31.
4 Paulson H, Bonini N, and Roth K. Polyglutamine disease and neuronal cell death. PNAS 2000; 97(24): 12957-58.
5 Semaka A, Collins J, Hayden M. Unstable familial transmissions of Huntington disease alleles with 27–35 CAG repeats (intermediate alleles). Am J Med Genet B Neuropsychiatr Genet 153: 314–320, 2010.
6 Wells R, Dere R, Hebert M, Napierala M, Son L. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Res 2005; 33(12): 3785–98.
7 Ranen N, Stine O, Abbott M, Sherr M, Codori A, Franz M, et al. Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am J Hum Genet 1995; 57: 593–602.
8 Pearson CE. Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol Med 2003; 9: 490–95.
9 Bates G. The molecular genetics of Huntington disease-a history. Nat Rev Genet. 2005; 6: 766-73.
10 Kremer B, Almqvist E, Theilmann J, Spence N, Telenius H, Goldberg P, Hayden R. Sex-Dependent Mechanisms For Expansions and Contractions of the CAG Repeat on Affected Huntington Disease Chromosomes. Am J Hum Genet 1995; 57: 343-50.
11 Potter N, Spector E, Prior T. Technical Standards and Guidelines for Huntington Disease Testing. Genet Med 2004; 6: 61-5.
12 Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, et al. Trinucleotide repeat length instability and age of onset in Huntington´s disease. Nat Genet 1993; 4: 387-92.
13 Brinkman R, Mezei M, Theilmann J, Almqvist E, Hayden M. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet 1997; 60: 1202–10.
14 Van Rij M, De Rademaeker M, Moutou C, Dreesen J, De Rycke M, Liebaers I. Preimplantation genetic diagnosis (PGD) for Huntington’s disease: the experience of three European centres. Eur J Hum Genet 2012; 20(4): 368-75.
15 Moore R, Xiang F, Monaghan J, Han D, Zhang Z, Edstrom L, et al. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet 2001; 69: 1385–8.
16 Margolis R, O’Hearn E, Rosenblatt A, Willour V, Holmes S, Franz M, et al. A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol 2001; 50: 373–80.
17 Wexler N. Huntington’s disease: advocacy driving science. Annu Rev Med. 2012; 63: 1-22.
18 Andrew S, Goldberg Y, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington´s disease. Nat Genet 1993; 4: 398-403.
19 Sánchez A, Castellvi-Bel S, Mila M, Genis D, Calopa M, Jiménez D,et al. Huntington´s disease: confirmation of diagnosis and presymptomatic testing in spanish families by genetic analysis. J Neurol Neurosurg Psychiatry 1996; 61: 625-7.
20 Alonso M, Yescas P, Cisneros B, Martínez C, Silva G, Ochoa A, et al. Analysis of the (CAG)n repeat causing Huntington´s disease in a Mexican population. Clin Genet 1997; 51: 225-30.
21 Lima e Silva T, Guerra H, Bertuzzo C, Lopes I. Molecular diagnosis of Huntington disease in Brazilian patients. Arq Neuropsiquiatr 2000; 58: 11-7.
22 Landles C, Bates G. Huntingtin and the molecular pathogenesis of Huntington disease. EMBO Rep 2004; 5(10): 958-63.
23 Wexler N, Lorimer J, Porter J, Gómez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. P Natl Acad Sci USA 2004; 101(10): 3498-503.
24 Gusella J, MacDonald M. Huntington’s disease: the case for genetic modifiers, Genome Medicine 2009, 1:80.
25 Kremer B, Almqvist E, Theilmann J, Spence N, Telenius H, Goldberg P, et al. Sex-Dependent Mechanisms For Expansions and Contractions of the CAG Repeat on Affected Huntington Disease Chromosomes. Am J Hum Genet 1995; 57: 343-50.
26 Gonzalez-Alegre P, Afifi A. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol 2006; 21: 223-9.
27 Stern R, Eldridge R. Attitudes of patients and their relatives to Huntington’s disease. J Med Genet 1975; 2(3): 217-23.
28 Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter J, Conneally P. Suicide risk in Huntington’s disease. J Med Genet 1993; 30: 293–295.
29 Paulsen J, Ferneyhough Hoth K, Nehl C, Stierman L. The Huntington Study Group. Critical Periods of Suicide Risk in Huntington’s disease. Am J Psychiatry 2005; 162: 725–31.
30 Clinical practice in medical genetics. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994;31:555-9.
31 Bernhardt C, Schwan A, Kraus P, Epplen J, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993-2004). Eur J Hum Genet 2009; 17: 295–300.
32 Harper P. Practical Genetic Counseling, 7th Ed., Oxford, Oxford University Press, 2010.

