










Serviços Personalizados
Journal

Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Medicina Interna
versão impressa ISSN 0250-3816versão On-line ISSN 1688-423X
Arch. Med Int vol.36 no. 1 Montevideo mar. 2014
Arch Med Interna 2014 - 36(1): 33-38
Caso clínico de interés
Colestasis intrahepática: un desafío diagnóstico
Intrahepatic cholestasis: a diagnostic challenge
Dr. Martín Collares
Res. Clínica Médica “A”. Hospital de Clínicas “Dr. Manuel Quintela”. Facultad de Medicina. UdelaR. Montevideo.
Dr. Marcelo Valverde
Prof. Adj. Clínica Médica “A”. Hospital de Clínicas “Dr. Manuel Quintela”. Facultad de Medicina. UdelaR. Montevideo.
Dra. Isabel Fernández
Prof. Adj. Clínica Médica “A”. Hospital de Clínicas “Dr. Manuel Quintela”. Facultad de Medicina. UdelaR. Montevideo.
Dra. Gabriela Ormaechea
Prof. Dir. Clínica Médica “A”. Hospital de Clínicas “Dr. Manuel Quintela”. Facultad de Medicina. UdelaR. Montevideo.
Centro de Trabajo: Clínica Médica “A” Prof. Dra. Gabriela Ormaechea. Hospital de Clínicas “Dr. Manuel Quintela”. Facultad de Medicina – Universidad de la República.
Correspondencia: Dr. Martín Collares. Gral. Urquiza 3040 apto. 311 - 2481 4958. Correo electrónico: martincollares@adinet.com.uy
Recibido: 16/04/13 - Aceptado: 14/03/14
Resumen:
La colestasis, término acuñado por el anátomo-patólogo Hans Pooper en 1956, constituye un verdadero síndrome clínico-analítico, que incluye un amplio espectro de entidades patológicas de severidad y pronóstico variable. Las manifestaciones clínicas principales de la colestasis derivan de la acumulación en plasma de sustancias normalmente excretadas en la bilis, vinculado a la disminución en la excreción biliar al tubo digestivo, base fisiopatológica de este síndrome. En su conjunto, las colestasis pueden clasificarse en intra o extrahepáticas, ictéricas o anictéricas y agudas o crónicas; asimismo, desde una visión etiológica, pueden catalogarse como congénitas, inflamatorias, mecánicas, tóxicas, metabólicas o neoplásicas. En el presente artículo, y a partir del reporte de un caso clínico, se analizan aspectos diagnósticos y terapéuticos del síndrome colestásico, proponiendo un algoritmo de actuación, de acuerdo a la mejor evidencia disponible a la fecha.
Palabras clave: Síndrome colestásico, Colestasis intrahepática, Colestasis Intrahepática Benigna Recurrente.
ABSTRACT:
Cholestasis, a term coined by Hans Pooper anatomic pathologist in 1956, is a real analytic clinical syndrome that includes a wide spectrum of pathological severity and outcome variable. Major clinical manifestations of cholestasis arising from the accumulation of plasma substances normally excreted in the bile, linked to decreased biliary excretion into the digestive tract, pathophysiological basis of this syndrome. Overall, cholestasis can be classified into intra or extrahepatic, icteric or anicteric and acute or chronic, also from an etiological view can be categorized as congenital, inflammatory, mechanical, toxic, metabolic, or neoplastic. In this article, and from clinical case report, we analyze the diagnostic and therapeutic cholestatic syndrome, proposing an algorithm of action, according to the best evidence available to date.
Keywords: Cholestatic syndrome, Intrahepatic cholestasis, Benign Recurrent Intrahepatic Cholestasis.
Introducción
La colestasis, término acuñado por el anátomo patólogo Hans Pooper en 1956, constituye un verdadero síndrome clínico-bioquímico, frecuente en la práctica clínica, que incluye un amplio espectro de condiciones patológicas de severidad y pronóstico variable(1). Conceptualmente se desarrolla frente a cualquier alteración que interfiera desde la síntesis biliar en el hepatocito y su modificación a nivel de los conductillos y ductos biliares, hasta su excreción a nivel del tubo digestivo. Por lo tanto, cualquier proceso que altere estos mecanismos fisiológicos producirá un complejo de manifestaciones clínicas, analíticas y anátomo-patológicas que se definen como colestasis(2).
El objetivo de la presente revisión es analizar conceptos básicos de la colestasis intrahepática como entidad desde el punto de vista clínico, diagnóstico y terapéutico, no incluyendo el análisis de las diferentes entidades responsables de la misma.
Caso clínico
Paciente de sexo masculino, 43 años de edad, sin antecedentes familiares, personales ni epidemiológicos a destacar. Consulta por cuadro clínico caracterizado por coloración amarillenta de piel y mucosas, prurito generalizado y astenia. De la enfermedad actual se destaca una historia de 15 días de evolución de coloración amarillenta de piel y mucosas, persistente y en aumento progresivo, acompañada de orinas hipercoloreadas e intenso prurito generalizado en cara, tronco y miembros, con compromiso palmoplantar, que por su severidad imposibilita conciliar el sueño. En forma concomitante refiere intensa astenia, anorexia y adinamia. Niega dolor abdominal de filiación biliar, no alteraciones del tránsito digestivo. El cuadro ha transcurrido en apirexia, sin otra sintomatología a destacar. Al examen físico se objetiva paciente lúcido, buen estado general, bien hidratado, apirético, con ictericia universal y verdínica, intenso prurito que motiva el rascado durante la valoración clínica, con lesiones secundarias él, sin anemia clínica ni lesiones hemorragíparas, no presenta dolor abdominal, no signos de Murphy ni de Bard y Pick, con examen cardiovascular, respiratorio y neurológico sin alteraciones.
De la analítica inicial se destaca: Hemoglobina 13,5 g/l; Plaquetas 220.000/mm3; Leucocitos 7.500/mm3 con fórmula conservada. Función renal, ionograma y glicemia normales. Hepatograma: Bilirrubina total 15,85 mg/dl, Bilirrubina directa 14,20 mg/dl; FA 438 UI/l; GGT 40 UI/l; TGP 200 UI/l; TGO 85 UI/l; Albúmina 3,83 g/l. Crasis sanguínea normal. Del resto de la analítica presenta un panel serológico viral no reactivo, tanto para virus hepatotropos (VHA, VHB, VHC) como para virus no hepatotropos (VEB, CMV). Presenta serologías para VIH y lúes no reactivas.
Frente a un paciente con la historia expuesta de ictericia, prurito y astenia, con alteraciones analíticas, donde se destaca una hiperbilirrubinemia a expensas de la bilirrubina directa, asociado a un aumento de la fosfatasa alcalina, se plantea el diagnóstico clínico-bioquímico de colestasis, destacando la ausencia de dolor abdominal de filiación biliar y fiebre. De la valoración imagenológica presenta a nivel de la ecografía abdominal, un hígado ecográficamente normal, sin dilatación de la vía biliar intra o extrahepática y sin lesiones ocupantes de espacio. En este punto, ante la indemnidad de la vía biliar, sin dilatación de la misma, se plantea el diagnóstico de colestasis intrahepática. En la evolución el paciente progresa desde el punto de vista sintomático, con aumento de la ictericia y del prurito, sin agregar nuevos síntomas o signos. Se inicia tratamiento sintomático del prurito en base a antihistamínicos, colestiramina y luego rifampicina, dada su persistencia a pesar de las medidas iniciales.
Para completar la valoración analítica se solicitaron anticuerpos antimitocondriales (AMA), antinucleares (ANA), antimúsculo liso (ASMA), y anticuerpos antiantígenos microsomales de hígado y riñón (anti LKM-1) que resultaron negativos. Con el objetivo de continuar con la valoración etiológica se solicitó colangiorresonancia magnética, la cual no evidenció dilatación universal ni quística de la vía biliar, así como tampoco imágenes de estenosis o tumores a dicho nivel. Dada la ausencia de diagnóstico etiológico de la colestasis se realizó punción biópsica hepática que evidenció parénquima hepático con retención biliar severa, compatible con colestasis colangiocelular, sin daño ductal ni ductopenia.
En un plazo aproximado de 6 a 8 semanas el paciente evoluciona clínicamente a la remisión espontánea del prurito y la ictericia, con franca mejoría del estado general y nutricional. De la evolución analítica se evidencia la peoría de los elementos de colestasis en concordancia con la clínica del enfermo (Tabla I). Se desataca que no mediaron medidas terapéuticas específicas más allá del tratamiento del prurito.

A las 12 semanas del inicio de los síntomas, en control ambulatorio, el paciente se encontraba asintomático, con examen clínico normal y sin alteraciones del hepatograma. Ante la presencia de un cuadro clínico, analítico e imagenológico compatible con una colestasis intrahepática, con evolución espontánea a la remisión, con valores de GGT persistentemente normales, se realiza el planteo diagnóstico probable de colestasis intrahepática benigna recurrente (CIBR), entidad incluida en el grupo de las colestasis hereditarias.
Discusión y comentarios
Desde el punto de vista clínico, el grupo de las colestasis se pueden clasificar en intra o extrahepáticas, ictéricas o anictéricas y agudas o crónicas; asimismo desde una visión etiológica pueden clasificarse en congénitas, inflamatorias, mecánicas, infecciosas, tóxicas, metabólicas o neoplásicas. La clasificación en colestasis intrahepática y extrahepática es la más aceptada dada su aplicabilidad clínica, siendo útil como punto de partida para la elaboración de los algoritmos diagnósticos universalmente aceptados y presentes en las guías internacionales.
La colestasis intrahepática es aquella que resulta de alteraciones a nivel hepatocelular, en los canalículos biliares o en los pequeños conductillos microscópicos (colestasis hepatocelular y colangiocelular) (Tabla II), mientras que la colestasis extrahepática es la originada por alteraciones de los conductos biliares macroscópicos. Es válido destacar que en algunas etiologías específicas existe solapamiento, con colestasis intra y extrahepática, como es el caso de la Colangitis Esclerosante Primaria o la Colangiopatía Isquémica(3). Desde el punto de vista clínico, la colestasis puede representar el problema clínico central del paciente (p. ej. cirrosis biliar primaria), mientras que en otros casos forma parte de un contexto clínico determinado que domina el cuadro (p. ej. injuria hepática en la sepsis). Las manifestaciones principales de la colestasis derivan de la acumulación en plasma de sustancias normalmente excretadas en la bilis, (ácidos biliares, bilirrubina y colesterol) y de la disminución en la excreción biliar al tubo digestivo, secundariamente pueden existir síntomas y/o signos derivados de la patología causal de la misma(4).
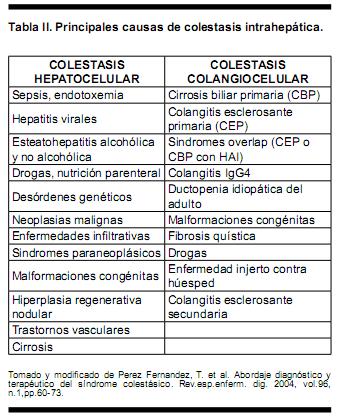
Es válido destacar que determinadas manifestaciones clínicas también variarán en función de la evolución aguda o crónica de la colestasis, así como también de su etiología intra o extrahepática. El prurito representa el problema clínico más frecuente, de intensidad variable, en ocasiones invalidante para el paciente y de difícil manejo terapéutico. El prurito de la colestasis afecta típicamente a palmas y plantas (manos y pies), pero puede ser generalizado. A diferencia de las enfermedades dermatológicas, no se objetivan lesiones cutáneas causantes del prurito, aunque en los casos graves pueden observarse lesiones secundarias al rascado, tales como excoriaciones o prurigo nodularis, en el que aparecen pápulas o nódulos hiperpigmentados debidos a cambios crónicos secundarios al rascado.
La ictericia representa otro de los signos clínicos característicos del síndrome colestásico, su intensidad y características clínicas son variables en función de la etiología y de la severidad de la colestasis. La presencia de xantomas y xantelasmas expresan el déficit en la excreción biliar del colesterol, siendo otro signo clínico frecuente en casos de colestasis de evolución crónica. En dichos casos se puede observar también la presencia de “clubbing” o deformación hipertrófica distal digital, osteoporosis secundaria, así como también manifestaciones disabsortivas vinculadas al déficit en la absorción de vitaminas liposolubles. Por último, en ocasiones pueden objetivarse síntomas y/o signos derivados de la etiología causal de la colestasis, tales como dolor abdominal, fiebre, síntomas constitucionales, manifestaciones autoinmunes sistémicas, etc.
En lo analítico la colestasis intrahepática presenta características bioquímicas particulares(2). Los hallazgos de laboratorio comunes a todas las formas de colestasis son el aumento de la fosfatasa alcalina (FAL), la gamma-glutamil-transpeptidasa (GGT), y la 5´nucleotidasa (que no forma parte del hepatograma de rutina). En ocasiones existe un aumento de la bilirrubina total (BT) a predominio de la bilirrubina directa (BD). Las transaminasas pueden estar aumentadas (ligera o marcadamente) o ser normales dependiendo de la etiología, siendo habitual en el patrón colestásico un incremento no mayor a 3 a 4 veces el límite superior de la normalidad.
La FAL corresponde a un grupo de enzimas distribuida en diversos sectores de la economía, como el hígado, hueso, riñón, intestino y placenta, pudiendo ser producida y secretada también por diferentes neoplasias. Las fracciones que se encuentran en el plasma son fundamentalmente la ósea, la hepática y, en menor escala, la intestinal. La fosfatasa alcalina placentaria se incrementa a partir del segundo trimestre y en especial durante el último trimestre del embarazo donde las cifras pueden ser del doble de lo normal. Valores elevados de FAL pueden observarse también en la infancia y adolescencia en relación a la intensa actividad osteoblástica en el contexto del crecimiento óseo. Su aumento en las enfermedades hepatobiliares no parece guardar relación con la disminución de la excreción de esta enzima por la bilis, por el contrario refleja un aumento en la síntesis hepática y su derivación hemática.
Una actividad sérica aumentada de la FAL puede preceder a la ictericia en procesos intra y extrahepáticos que afectan la función excretora del hígado. Cuando no hay otros indicios de enfermedad hepática es importante establecer el origen de una elevación de la FAL. Se han propuesto diversas opciones como el estudio de la labilidad térmica, la investigación de las isoenzimas de la FAL, o la medición de la actividad de otras enzimas de membrana como la GGT y la 5`nucleotidasa.
Estas últimas son las más utilizadas. La GGT es una enzima que se encuentra en las membranas celulares de diversos órganos como hígado, riñón, páncreas, bazo, corazón, cerebro y vesículas seminales. La actividad sérica de esta enzima se encuentra elevada en enfermedades hepáticas, biliares y pancreáticas. Su elevación en enfermedades hepatobiliares aumenta en forma paralela a la FAL pero es de mayor sensibilidad que ésta; también aumenta en casos de inducción de la actividad del citocromo P-450 por alcohol o drogas. Un aumento de la GGT o de la 5`nucleotidasa sugiere el origen hepático de una FAL elevada ya que la GGT no aumenta en pacientes con enfermedades óseas. Cuando el flujo biliar cesa en forma completa las cifras de bilirrubina se estabilizan en una meseta entre 20 y 30 mg/dl. El aumento de la BD es una manifestación analítica frecuente en la colestasis, de severidad variable y de menor sensibilidad diagnóstica que otras determinaciones como las elevaciones de la GGT y FAL, sabiendo que existen formas de colestasis anictéricas, así como casos en los cuales la hiperbilirrubinemia es de aparición tardía.
Algoritmo diagnóstico
Es válido jerarquizar previo al análisis de los estudios bioquímicos e imagenológicos la importancia de la realización de una anamnesis y exámen físico detallados, debiendo hacer énfasis en aquellos antecedentes, síntomas y/o signos que orienten a una etiología extrahepática de la colestasis, así como una detallada historia que incluya el consumo de fármacos, hierbas y yuyos en los 6 meses previos(5). Se destacan asimismo algunos subgrupos de pacientes con condiciones clínicas donde la colestasis intrahepática es de relativa frecuencia como el embarazo, la infección por VIH, el trasplante de médula ósea o de órgano sólido, etc. Estos elementos clínicos pueden ofrecer una orientación diagnóstica inicial para guiar la solicitud de exámenes complementarios en forma lógica y secuencial, así como la adopción de medidas terapéuticas iniciales. En función de la clasificación mencionada en colestasis intrahepática y extrahepática, se expone un algoritmo diagnóstico propuesto en las guías y protocolos internacionales (Figura 1).
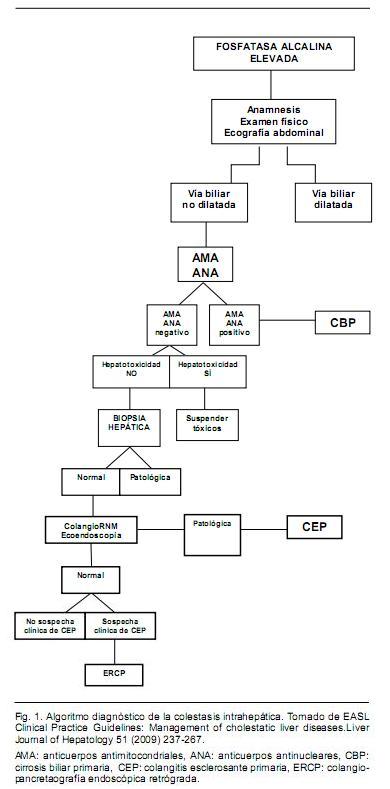
El primer escalón diagnóstico será identificar si existe o no dilatación de la vía biliar extrahepática. En tal sentido, en razón de su accesibilidad, costo, no invasividad, sensibilidad y especificidad diagnóstica para valorar la vía biliar, la ecografía abdominal se impone como el primer procedimiento diagnóstico(6). La tomografía computada (TC) de abdomen no aporta ventajas en esta instancia(7). Si la ecografía no evidencia una obstrucción mecánica biliar, se debe abordar el algoritmo diagnóstico de las colestasis intrahepáticas.
En primer término se deben solicitar los títulos de anticuerpos antimitocondriales (AMA) y anticuerpos antinucleares (ANA) para descartar el diagnóstico de cirrosis biliar primaria (CBP), que constituye la causa más frecuente de obstrucción de la vía biliar intrahepática. De obtener negatividad para dichos anticuerpos, y de acuerdo al algoritmo diagnóstico expuesto, una vez descartada la posibilidad de hepatotoxicidad se debe proceder a la biopsia hepática. Con respecto a esta herramienta diagnóstica, se recomienda una muestra histopatológica representativa a aquella que cumple con los siguientes criterios: 10 mm de longitud, 1,4 mm de ancho, y al menos 11 espacios porta (8). De no evidenciar signos anátomo-patológicos indicativos de colestasis, se debe valorar imagenológicamente la vía biliar. Si bien la colangiopancreatografía retrógrada endoscópica (ERCP) constituye el gold standard para detectar la obstrucción del árbol biliar, aun en centros experimentados tiene una tasa de complicaciones no despreciable que oscila entre el 3 y 5%; por tal razón cobran jerarquía otros procedimientos diagnósticos como la colangiorresonancia magnética (colangio-RM) o la ecoendoscopia biliar(9,10). Por tanto, en primer término, se debe solicitar una colangio-RM, opción segura para valorar el árbol biliar y tan eficaz como la ERCP(11), siendo la ecoendoscopía biliar otra opción diagnóstica comparable a la colangio-RM en centros experimentados.
De todos modos, de existir una firme sospecha clínica de CEP sin hallazgos característicos en la colangio-RM o ecoendoscopia biliar, se impone la ERCP como el siguiente procedimiento diagnóstico. Una vez descartada esta posibilidad, y si la situación clínica lo permite, debe establecerse un compás de espera respecto a los procedimientos diagnósticos, revalorando al paciente desde el punto de vista clínico, analítico e imagenológico de acuerdo con las características evolutivas del cuadro.
Manejo terapéutico
El síndrome colestásico da lugar a una variada signo-sintomatología que en ocasiones amerita la adopción de conductas terapéuticas sintomáticas, en virtud de la relevancia clínica de dichas manifestaciones. El tratamiento específico va a depender de la etiología de la colestasis, que varía desde la simple suspensión de determinados fármacos potencialmente responsables de la colestasis hasta el trasplante hepático, pasando por el uso de tratamientos inmunosupresores o procedimientos quirúrgicos sobre la vía biliar. En relación al manejo específico del prurito colestásico se debe destacar que en un porcentaje no despreciable de casos constituye un síntoma invalidante y de abordaje terapéutico complejo. La etiología del prurito colestásico no es del todo clara, pudiendo incluir mecanismos neurohumorales tanto centrales como periféricos(12).
El tratamiento propuesto por las guías internacionales se basa en la utilización de diversos recursos terapéuticos (Tabla III). El primer paso consiste en la administración de colestiramina, resina de intercambio aniónico que liga las sales biliares en la luz intestinal, bloqueando su absorción(13,14).Se suele prescribir a una dosis inicial de 4 g/día, pudiendo incrementarla según la respuesta hasta 16 g/día.

El fármaco suele ser efectivo a partir del tercer día del inicio del tratamiento. Se recomienda un intervalo de al menos 4 horas entre la ingesta de colestiramina y de cualquier otro fármaco que también pueda ser captado por la resina, especialmente ácido ursodesoxicólico (AUDC) y anticonceptivos hormonales. Si las resinas son ineficaces para el control del prurito, el siguiente paso consiste en la adminis-tración de rifampicina, antibiótico con propiedades de inducción enzimática, que puede administrarse durante periodos prolongados a dosis de 10 mg/kg/d, aunque pueden ser suficientes dosis menores a 300 mg/día(15,16). Como existe un riesgo potencial de hepatotoxicidad se recomienda un control periódico de las transaminasas. La rifampicina, además de aliviar el prurito también produce una nota-ble reducción de la colestasis, especialmente en los niveles de FA y GGT. Cuando no existe respuesta terapéutica a los anteriores agentes puede utilizarse la naltrexona, como antagonista no selectivo de los opioides endógenos. Este fármaco, a dosis de 50 mg/día sería de utilidad para el tratamiento del prurito, mejorando también los síntomas depresivos asociados a las colestasis crónicas, principalmente a la CBP.
Hay datos sobre la potencial efectividad de la sertralina, inhibidor selectivo de la recaptación de serotonina tratamiento de primera línea del prurito colestásico(17). Sin embargo la efectividad parecer ser menor, y también acompañada de una elevada tasa de efectos secundarios. En casos de prurito refractario a los anteriores tratamientos se ha mostrado muy eficaz la utilización de sistemas de soporte hepático artificial(18). En un estudio publicado recientemente se ha demostrado la eficacia de esta técnica utili-zando el sistema MARS® en una serie de 20 pacientes con prurito refrac-tario. Otros procedimientos de diálisis con albúmina, así como plasmaféresis o derivación externa de la vía biliar también se han utilizado como procedimientos alterna-tivos con experiencia limitada. Finalmente, el trasplante hepático constituye la última opción terapéutica en casos de prurito severo refractario(19). Si bien no constituye una indicación absoluta, sí es una opción válida en pacientes con prurito intratable.
En dichos casos se solicita la adjudicación de un MELD por vía de excepción. La astenia y la fatiga constituyen otro problema clínico frecuente que amerita un abordaje individualizado. Se deben descartar otros problemas clínicos concomitantes que puedan generar esta sintomatología (anemia, hipotiroidismo, diabetes mellitus, depresión, etc.) y tratarlos oportunamente. No existen tratamientos eficaces para la astenia colestásica, y el trasplante hepático no es una opción válida en ausencia de otras indicaciones formales. El modafinilo se ha mostrado eficaz en algunos estudios clínicos, fundamentalmente en casos de somnolencia diurna excesiva(20). Los pacientes con colestasis presentan un riesgo incrementado de osteoporosis, este riesgo es aún mayor en aquellos pacientes con colestasis severa y en hombres. En relación al tratamiento de la osteoporosis en este escenario, se recomienda la suplementación con vitamina D, calcio y la utilización de bifosfonatos(21).
Asimismo es recomendable la administración de vitaminas liposolubles (A, E, K) en caso de esteatorrea, existiendo evidencia a favor de la utilización de vitamina K por vía parenteral previo a la realización de procedimientos invasivos. Todas las enfermedades colestásicas crónicas tienen como complicación en su evolución el desarrollo de dislipemias, con su máxima expresión en la CBP(22). De todos modos no se ha demostrado un incremento en el riego cardiovascular absoluto en estos pacientes. En estos casos el tratamiento con ácido ursodesoxicólico (AUDC) permite disminuir las cifras de LDL colesterol(22). De todas formas, en caso de existir un riesgo cardiovascular incrementado, historia familiar de cardiopatía isquémica o antecedentes personales de enfermedad vascular, el empleo de estatinas y de fibratos es seguro, inclusive en aquellos pacientes con hepatograma alterado(23,24).
Por su parte, la Colestasis Intrahepática Benigna Recurrente (CIBR) es una enfermedad caracterizada por episodios recurrentes y autolimitados de ictericia y prurito intensos, de duración variable de semanas a meses(25). La mayoría de los casos descritos son esporádicos, pero hasta en un 50% se demuestra una historia familiar de colestasis, con un patrón de herencia autosómico recesivo de la enfermedad. Se ha logrado ubicar el defecto genético a nivel del brazo largo del cromosoma 18, gen que traduce una ATPasa (ATP8B1) que participa en la translocación de aminofosfolípidos a nivel del hepatocito, paso necesario para la secreción de sales biliares(1).
El gen ATP8B1 se localiza en el cromosoma 18q21-q22 y codifica una ATPasa de tipo P, llamada FIC1. Dicha ATPasa es una proteína integral de membrana, miembro de la familia de las ATPasas tipo P, subfamilia P4, y desempeña un importante rol en el transporte de aminofosfolípidos a través de la membrana celular. En la Figura 2 se esquematizan los mecanismos moleculares principales involucrados en las colestasis hereditarias más frecuentes, haciendo especial énfasis en el mecanismo de la CIBR. Con respecto a la presentación clínica, el debut de la enfermedad ocurre típicamente durante la adolescencia o hasta la tercera década de la vida.
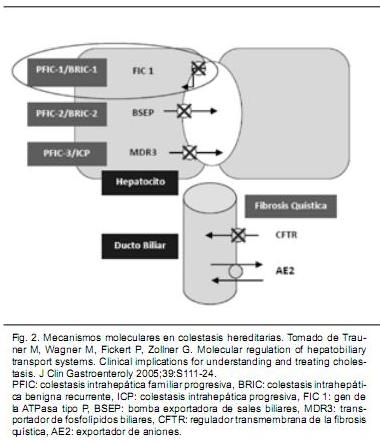
Cada evento colestásico puede durar de semanas a meses con una media de aproximadamente 90 días. Asimismo, los períodos libres de síntomas varían de meses a años(26,28). Los exámenes de laboratorio, durante los episodios sintomáticos revelan un patrón clásicamente colestásico, con una elevación de la fosfatasa alcalina de al doble del límite superior de la normalidad. La hiperbilirrubinemia es a predominio de la bilirrubina directa y puede elevarse a más de 10 veces el valor normal.
Las transaminasas cursan habitualmente con valores normales o discretamente elevados. La característica que distingue a esta entidad de otras formas de colestasis intrahepática (incluyendo las formas familiares) es el nivel de GGT, el cual permanece normal o ligeramente elevado. En nuestro paciente este dato fue relevante para la orientación diagnóstica(27).
Los criterios diagnósticos de CIBR son los siguientes(1):
· Al menos dos episodios de ictericia separados por un intervalo libre (de meses a años).
· Analítica concordante con colestasis intrahepática.
· GGT normal o discretamente elevada.
· Prurito severo secundario a colestasis.
· Histología hepática con colestasis centrolobular.
· Vía biliar intrahepática y extrahepatica normal, demostrado por colangiografia o colangio-RNM.
· Exclusión de otras causas de colestasis.
La evolución natural de la CIBR es benigna, sin progresión a fibrosis o insuficiencia hepática. Es válido destacar dentro de este heterogéneo conjunto de entidades constituido por las colestasis hereditarias a las colestasis familiares progresivas. En nuestro caso puntual ciertas características clínicas, cronológicas y analíticas nos alejan de este planteo diagnóstico, resaltando la utilidad de los estudios genéticos a través de la identificación de los denominados “genes colestásicos”(29).
Discusión y Comentarios
Se ha presentado el caso clínico de un paciente portador de una colestasis intrahepática benigna recurrente, entidad incluida en el heterogéneo grupo de las colestasis hereditarias. A punto de partida de dicho caso se realizó una revisión bibliográfica acorde a una patología de relativa frecuencia en la práctica clínica habitual, considerada un verdadero síndrome, que incluye una gran diversidad de etiologías con sus particularidades clínicas, bioquímicas, imagenológicas y terapéuticas. Se ha hecho especial énfasis en el algoritmo diagnóstico de la colestasis intrahepática, abordando finalmente los aspectos terapéuticos cardinales de este síndrome, sin profundizar en el tratamiento específico de cada entidad “per se”.
BIBLIOGRAFÍA
1.Elias E, Boyer JL, Mechanisms of Intrahepatic Cholestasis. En Progress in Liver Diseases. Popper H, Schaffner F. ed Grune & Stratton N. York 1979; vol VI; 667-680.
2.Kaplan MM. In Diseases of the Liver. Schiff L. and Schiff. ER. ed J.B. Lippington Co. Philadelphia, USA, 1993:223-250.
3.Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5: 776-782.
4.Leevy CM, Sherlock S, Tygstrup N, Zetterman R. Standardization of Nomenclature, Diagnostic Criteria, and Prognosis. En Diseases of the Liver and Biliary Tract. ed. Comm Raven Press, N. York 1994; 339-400.
5.Simon F. Drug induced Cholestasis: Pathobiology and clinical features. Clinics in Liver Disease 1998; 483-499.
6.Pedersen OM., Nordgârd K., Kvinnsland S. Value of Sonography in Obstructive Jaundice. Limitations of Bile Duct Caliber as an Index of Obstruction. Scand. J. Gastroenterol 1987; 975-981.
7.Baillie J, Poulson EK, Vitellas KM. Biliary imaging: a review. Gastroenterology, 6: 1686-1699.
8.Standish RA, Cholongitas E, Dhillon A, Burroughs AK, Dhillon AP. An appraisal of the histopathological assessment of liver fibrosis. Gut 2006; 55: 569-578.
9.Freeman ML, Nelson DB, Sherman S, Haber GB, Herman ME, Dorsher PJ, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335: 909-918.
10.Maggs JR, Chapman RW. An update on primary sclerosing cholangitis. Curr Opin Gastroenterol 2008; 24: 377-383.
11.Scheuer PJ. Primary biliary cirrhosis: diagnosis, pathology and pathogenesis. Postgrad Med J 1983; 59: 106-115.
12.Kremer AE, Beuers U, Oude-Elferink RP, Pusl T. Pathogenesis and treatment of pruritus in cholestasis. Drugs 2008; 68: 2163-2182.
13.Datta DV, Sherlock S. Cholestyramine for long term relief of the pruritus complicating intrahepatic cholestasis. Gastroenterology 1966; 50:323-332.
14.Rust C, Sauter GH, Oswald M, Buttner J, Kullak-Ublick GA, Paumgartner G, et al. Effect of cholestyramine on bile acid patterns and synthesis during administration of ursodeoxycholic acid in man. Eur J Clin Invest 2000; 30:135-139.
15.Khurana S, Singh P. Rifampicin is safe for the treatment of pruritus die to chronic cholestasis: a meta-analysis of prospective randomized-controlled trials. Liver Int 2006; 26:943-948.
16.Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile acid binding agents, opioid antagonists or rifampicin in the treatment of cholestasis-associated pruritus. Am J Gastroenterol 2007; 102:1528-1536.
17.Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45: 666-674.
18.Pares A, Cisneros L, Salmeron JM, Caballeria L, Mas A, Torras A, et al. Extracorporeal albumin dialysis: a procedure for prolonged relief of intractable pruritus in patients with primary biliary cirrhosis. Am J Gastroenterol 2004; 99:1105-1110.
19.Gross CR, Malinchoc M, Kim WR, Evans RW, Wiesner RH, Petz JL, et al. Quality of life before and after liver transplantation for cholestatic liver disease. Hepatology 1999; 29: 356-364.
20.Newton JL, Gibson JG, Tomlinson M, Wilton K, Jones DEJ. Fatigue in primary biliary cirrhosis is associated with excessiveday time somnolence. Hepatology 2006; 44:91-98.
21.Pares A, Guanabens N. Osteoporosis in primary biliary cirrhosis: pathogenesis and treatment. Clin Liver Dis 2008;12:407-424.
22.Allocca M, Crosignani A, Gritti A, Ghilardi G, Gobatti D, Caruso D, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795-1800.
23.Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology 2005; 41:690-695.
24.Nakamuta M, Enjoji M, Kotoh K, Shimohashi N, Tanabe Y. Long-term fibrate treatment for PBC. J Gastroenterol 2005; 40:546-547.
25.Luketic A, Shiffman M. Benign recurrent intrahepatic cholestasis. Clin Liver Dis 2004; 133-149.
26.Brenard R, Geubel AP, Benhamou JP. Benign recurrent intrahepatic cholestasis. J Clin Gastroenterol 1989; 11:546-551.
27.Van Ooteghem NA, Klomp LW, Van Bergehenegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familiar intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J. Hepatol. 2002; 36:439-443.
28.Hernández Nelia. Colestasis intrahepática recurrente benigna.Archivos de Medicina Interna 2002; V24, N4: 135-137.
29.Chiodi D, Berrueta J, Labandera D, López C, Hernández N. Ayer: Colestasis intrahepática familiar progresiva tipo 3. Hoy colestasis ABCB4/MDR3. Carta Gastroenterológica 2010; vol 17 (Nº 2) 25-48.

