











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
La aplasia cutis (AC) es un defecto congénito poco frecuente, con una incidencia de 0,5-3/10.000 recién nacidos1,2. Fue descripta en 1767, por primera vez, por Cordon3, en un caso de AC en miembros inferiores.
Se localiza más frecuentemente en el cuero cabelludo, donde se asocia a defecto óseo en el 20% de los casos1. Constituye una lesión de tipo no inflamatoria y bien delimitada, de extensión variable, desde 0,5 a 10 cm o más. La mayoría de los episodios son esporádicos, pero se han descripto casos familiares. Puede asociarse a otras anomalías físicas, incluso formando parte de síndromes malformativos o cromosomopatías. El pronóstico depende de la profundidad del defecto.
Caso clínico
Recién nacido de sexo femenino, segunda gestación. Embarazo bien controlado, no consumo de fármacos o drogas. Amenaza de parto de pretérmino a las 36 semanas, realizándose diagnóstico ecográfico de microcefalia con encefalocele parietooccipital derecho, solución de continuidad de 9 mm, oligoamnios y arteria umbilical única. Se inicia abordaje interdisciplinario coordinado con equipo de cuidados paliativos. Cesárea a las 38 semanas, por disminución de movimientos fetales, presentación cefálica. Recién nacido vigoroso, peso al nacer 2660 g, longitud 43 cm. Perímetro cefálico 30 cm, microcefalia, hipoplasia ósea y cutánea, encefalocele en línea media de cráneo de 5 x 5 cm cubierto por aracnoides, con áreas hemorrágicas y arteria umbilical única, sin otras malformaciones a destacar (Figura 1).
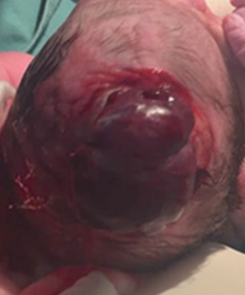
Figura 1 Recién nacido con hipoplasia ósea y cutánea, encefalocele en línea media de cráneo de 5 x 5 cm cubiertos por aracnoides con áreas hemorrágicas.
Sin antecedentes familiares a destacar.
Ingresa a cuidados intensivos neonatales, hemodinámicamente estable, ventilando al aire. Defecto cubierto por gasa estéril con suero tibio y curación plana con compresa. Se coloca acceso vascular periférico y central. El equipo de neurocirujanos y cirujanos plásticos realizan curación del defecto con amnios y compresa.
Resonancia magnética de cráneo: confirma defecto parasagital parietal derecho, con compromiso del parénquima y estructuras vasculares arteriales. Disminución volumétrica del hemisferio cerebral derecho, rotación de estructuras hacia la brecha, comprometiendo parénquima cerebral, cuerpo del ventrículo lateral y ganglios de la base. Lateralización de línea media hacia derecha (Figura 2).
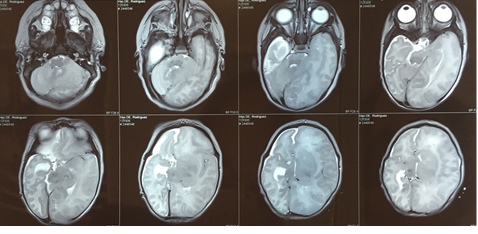
Figura 2 Resonancia magnética de cráneo observa defecto parasagital parietal derecho, con compromiso del parénquima, disminución volumétrica, rotación de estructuras hacia la brecha y desviación de línea media.
Angiografía y venograma de cráneo: ascenso de la bifurcación de la carótida derecha, elevación de la arteria cerebral anterior y cerebral media derechas. Arteria cerebral media en posición vertical. Valoración venosa: adecuada topografía y señal del flujo del seno longitudinal superior, así como del seno recto y venas cerebrales internas, estas últimas ascendidas. Fosa posterior hipoplasia de seno transverso izquierdo (Figura 3).
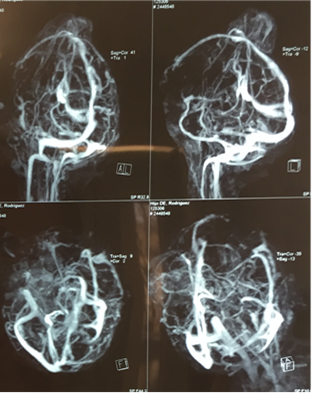
Figura 3 Angiografía y venograma de cráneo, se visualiza la alteración anatómica con mal posición de los vasos.
Ecocardiografía sin cardiopatía estructural. Cariotipo normal.
Al cuarto día de vida, se realiza plastia de duramadre con amnios y cierre de piel, sin incidentes. Ingresa a centro de tratamiento intensivo ventilado, estable clínicamente, analgesia con fentanyl y dipirona. Se extuba en el posoperatorio inmediato, CPAP por tres días y luego se mantiene ventilando al aire. A los nueve días de vida, paciente estable, punto de sutura dehiscente que evoluciona a la peoría. Importante protrusión de contenido encefálico edematoso a los 14 días de vida, por lo cual se decide nueva plastia con colgajo de fascia lata, resección de tejido encefálico necrótico y cierre de piel. Concomitantemente, presenta múltiples hiponatremias mantenidas, requiriendo varias correcciones. A las 24 horas del posoperatorio el colgajo evoluciona a la necrosis, dada la mala evolución anterior, se realiza planificación de cuidados con adecuación de tratamientos en conjunto con los padres. Se ajusta tratamiento analgésico multimodal.
Se acepta una tercera intervención dada la necesidad de mejorar la calidad de vida. A los 27 días, plastia de fascia lata y colgajo cutáneo que requiere cierre por segunda intención en gran parte de su extensión, colocando parches de hidrocoloides (Figura 4). A los 31 días de vida, episodio de movimiento tónico clónico y parpadeo, por lo que se plantea episodio convulsivo, se inicia fenobarbital y no reitera.
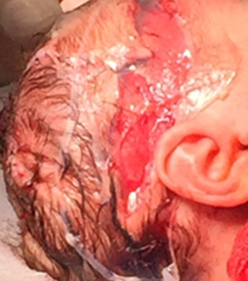
Figura 4 Recién nacido, 27 días, luego de tercera intervención quirúrgica donde se realiza plastia de fascia lata y colgajo cutáneo.
Buena evolución luego de 39 días de internación, con alimentación enteral total y un peso de 3090 g. Se decide otorgar alta, con curaciones diarias evitando la manipulación innecesaria de la zona afectada, utilizando parches de hidrocoloides.
Se mantiene analgesia a base de licor de brompton, ibuprofeno y dextrosa. Seguimiento en domicilio por equipo de cuidados paliativos.
La evolución fue un crecimiento aceptable, retraso psicomotor. Cierre de colgajo por segunda. Varias internaciones por vómitos y depresión de conciencia vinculados a hiponatremias en estudio y en tratamiento con complemento de sodio vía oral, con planteo de encefalopatía perdedora de sal.
Discusión
La AC congénita es una rara alteración caracterizada por la ausencia o el adelgazamiento de la epidermis, dermis, y, en ocasiones, de los tejidos subyacentes1, como presentó nuestra paciente.
Su incidencia es de 0,5 a 3 cada 10.000 recién nacidos vivos, según distintos reportes, siendo más frecuente en el sexo femenino (7:5). Un 80% a 90% presenta las lesiones típicas1,2,4. La literatura describe que 20% asocia un defecto óseo1,2, pero no se cuenta con la incidencia de las formas graves en las que además del defecto, se asocian alteraciones de tejidos subyacentes, como en nuestro caso, siendo rara su asociación con encefalocele.
Existen numerosas hipótesis sobre su etiopatogenia, desde la adhesión de bandas amnióticas, infecciones intrauterinas, ruptura de la piel por la tensión que provoca el rápido crecimiento cerebral o la asociación con distintos fármacos o drogas de abuso (metimazol, misoprostol, cocaína o alcohol). Ninguna se ha confirmado. Sin embargo, se han sugerido factores predisponentes como traumatismos intrauterinos, exposición a radiación y factores genéticos. Algunos autores plantean que un defecto del cierre del tubo neural podría ser responsable, describiendo casos de AC con valores altos de alfafetoproteína y acetilcolinesterasa positiva en líquido amniótico5-7.
También se ha descripto su asociación con síndromes, como el de Adams-Oliver, trisomía 13, 12p, hipoplasia dérmica focal, entre otros, y puede estar vinculada a patologías cardiovasculares, del sistema nervioso central, oculares, del aparato locomotor, cutáneas y labio leporino1,8.
En nuestro caso no encontramos asociación con ninguna de las posibles causas antes mencionadas, y, dada la ausencia de afectación de otras estructuras, no se asoció con ningún síndrome.
Existen dos clasificaciones. La de Frieden de 1986, la más utilizada con nueve grupos, y la de Sybert, con cinco grupos. En ellas se relacionan características del defecto cutáneo, ausencia o presencia de malformaciones asociadas y herencia genética9-12 (Tablas 1 y 2). Nuestro paciente se encontraría en el grupo 1 de Frieden y en el grupo I de Sybert, ya que la AC se encuentra solo en cuero cabelludo, no asocia síndromes y se plantea que el encefalocele es consecuencia de la ausencia de tejido óseo.
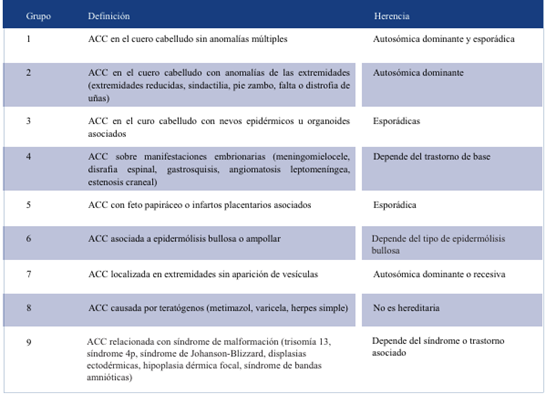
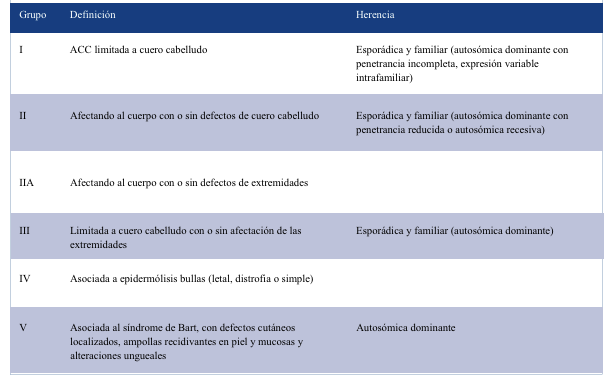
El aspecto de las lesiones es diverso, pueden variar en tamaño, forma y región. Frecuentemente, en 80%-90% de los casos, se trata de un defecto único, de tipo no inflamatorio, bien delimitado, oval o circular, de 2 cm de diámetro. Localizado en la línea media corporal, principalmente en el cuero cabelludo. En este caso, si bien el defecto presenta algunas características clásicas, su tamaño, con gran afectación de tejido óseo asociando el encefalocele, lo destaca de las presentaciones más frecuentes.
Desde el punto de vista clínico se clasifica en membranosa (la más frecuente), irregular o estrellada, asociada a alteraciones embriológicas de órganos internos y con ausencia congénita de la piel13,14. Este caso lo clasificamos como irregular o estrellada: las lesiones son de mayor tamaño, suelen estar erosionadas al nacimiento, se localizan en el cuero cabelludo, interparietal y a menudo se asocian a un defecto óseo. En los casos de alteraciones óseas importantes, la duramadre está expuesta y suele estar adelgazada o ausente13.
El diagnóstico es clínico, la histología no es de rutina. Existen casos con sospecha diagnóstica intrauterina, por ecografías, confirmándose al nacimiento.
En nuestra paciente debemos realizar el diagnóstico diferencial con encefalocele por defecto del cierre del tubo neural. Nos aleja de este diagnóstico las características de la lesión en saca bocado, y que no se haya detectado en ecografías previas las características de la imagen en la resonancia, nos lleva a pensar que el encefalocele se formó después del cierre del tubo neural. El contenido encefálico se encontraba intacto, esto también nos aleja de este planteo, dado que cuanto más tiempo se encuentra expuesto el encéfalo al líquido amniótico intraútero, irá destruyéndose hasta desaparecer.
La AC es una patología benigna, pero en los casos severos se han descripto complicaciones hemorrágicas (por compromiso del seno sagital superior) e infecciosas como meningitis, comprometiendo la vida del recién nacido. El tratamiento de la AC depende de la región corporal afectada, la profundidad y la extensión. Las lesiones pequeñas y superficiales tienden a cerrar espontáneamente en semanas o meses, dejando una cicatriz atrófica y sin pelo9.
En los defectos grandes a nivel de cuero cabelludo, sobre todo los que asocian defectos óseos, se recomienda la restauración temprana del defecto óseo y cutáneo en una única intervención en la que se realizaría una craneoplastia utilizando material óseo autógeno9,12,15. Para el defecto cutáneo se puede recurrir a colgajos cutáneos, expansiones o injertos de piel. La cobertura precoz del defecto no solo disminuye el riesgo vital, sino que además reduce significativamente las complicaciones locales y facilita la reconstrucción definitiva9. En nuestro caso, para el cierre por segunda intención se utilizaron parches de hidrocoloides, ya que mantienen la herida aislada del exterior y por medio del contacto con el exudado de la lesión forma un gel viscoso manteniendo un ambiente húmedo, favoreciendo la cicatrización y evitando la infección al proporcionar una barrera antibacteriana. Estos pueden mojarse, no producen dolor al ser retirados, favorecen el aporte de oxígeno y nutrientes a través de la angiogénesis, facilitan la migración celular, disminuyen el tiempo y el número de las curaciones locales y mejoran los resultados estéticos16.
En conclusión, un adecuado diagnóstico prenatal permite coordinar el nacimiento en una institución que pueda promover atención temprana, adecuada e individualizada al recién nacido.
El equipo interdisciplinario para el diagnóstico y tratamiento oportuno es uno de los pilares fundamentales para el manejo de los casos de AC, como el nuestro.
Si bien la AC es una patología con una baja incidencia, siendo más rara aun la asociación con encefalocele, es necesario resaltar el conocimiento generado en su diagnóstico y manejo dadas las potenciales complicaciones que pueden llevar a una elevada mortalidad.

