










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versão impressa ISSN 0004-0584versão On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.87 no.4 Montevideo dez. 2016
Asociación de melanocitosis dérmica con enfermedades lisosomales
Association of dermal melanocytosis with lysosomal storage diseases
Lucía Cibils1, Conrado Medici2, Francisco Espinel3, Karina Malan4, Aida Lemes5, Alfredo Cerisola6, Gabriel Gonzalez71. Asistente Neuropediatría. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
2. Prof. Adj. Neuropediatría. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
3. Ex-Posgrado Neuropediatría. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
4. Residente pediatría. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
5. Genética médica. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
6. Prof. Agdo. Neuropediatría. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
7. Prof. Neuropediatría. Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
Cátedra de Neuropediatría. Facultad de Medicina. UDELAR. CHPR.
Trabajo inédito.
Declamamos no tener conflictos de intereses.
Fecha recibido: 2 de marzo de 2016.
Fecha aprobado: 27 de abril de 2016.
Resumen
Introducción: la melanocitosis dérmica incluye un espectro de lesiones de piel que abarca la mancha mongólica, entre otras lesiones. Las enfermedades lisosomales son afecciones de base genética que se caracterizan por la acumulación de metabolitos insolubles parciamente degradados en los compartimentos lisosomales, debido a una determinada deficiencia enzimática. Las deficiencias de b-galactosidasa y de a-L-iduronidasa provocan la gangliosidosis GM1 y la mucopolisacaridosis tipo I respectivamente, ambas presentando similitudes en su presentación clínica. La asociación de la melanocitosis dérmica con las enfermedades lisosomales es poco común y mal entendida.
Objetivo: reportar dos pacientes con esta rara asociación.
Casos clínicos: dos varones de 3 y 9 meses sin antecedentes prenatales ni perinatales a destacar y antecedentes de infecciones respiratorias reiteradas. Se presentaron con retraso del desarrollo, hipotonía central y trastorno deglutorio. Al examen se constató hepatomegalia, fascies tosca y melanosis dérmica extensa. Los estudios permitieron diagnosticar al paciente de 3 meses mucopolisacaridosis Tipo I y al de 9 meses gangliosidosis GM1.
Discusión: no se conoce exactamente la causa de esta asociación. Se plantea que sería el resultado de la acumulación de gangliósidos y heparán sulfato que estimularían al receptor del factor de crecimiento neuronal de tipo tirosinquinasa, deteniendo la migración de los melanocitos en la dermis. Por lo tanto la melanosis dérmica aberrante, en el contexto clínico adecuado, puede ser un signo que facilite el diagnóstico de una enfermedad lisosomal subyacente.
Palabras clave:
MELANOCITOSIS DÉRMICA
ENFERMEDADES POR ALMACENAMIENTO LISOSOMAL
GANGLIOSIDOSIS GM1
MUCOPOLISACARIDOSIS I
Summary
Introduction: dermal melanocytosis includes a spectrum of skin lesions, mongolian spots being one of them. Lysosomal storage diseases are characterized by the accumulation of partially degraded insoluble metabolites in lysosomal compartments due to enzyme deficiency. Deficiency in b-galactosidosisis is the cause of GM1 gangliosidosis and deficiency in a-L-iduronidasa of mucopolysaccharidosis type I. Both have similar clinical presentations. Association of dermal melanocytosis and lysosomal storage diseases is uncommon and misunderstood.
Objective: to report the case of two patients with this rare association.
Clinical cases: the study presents two boys, 3 and 9 months old, with no remarkable family, pregnancy or delivery history. Both had repeated respiratory tract infections. They presented with developmental delay, central hypotonia and swallowing disorder. Upon clinical examination they showed hepatomegaly, coarse facies and extensive dermal melanocytosis. They were diagnosed with GM1 gangliosidosis and mucopolysaccharidosis type I.
Discussion: the cause of this association is not well known. It is hypothesized that accumulation of gangliosides and heparan sulfates stimulates tyrosine-kinase neuronal growth factor receptor, stopping dermal melanocytosis migration. Therefore extensive dermal melanocytosis, in an appropriate clinical setting, may contribute to diagnosing lysosomal storage diseases.
Key words:
DERMAL MELANOCYTOSIS
LYSOSOMAL STORAGE DISEASES
GANGLIOSIDOSIS GM 1
MUCOPOLYSACCHARIDOSIS I
Introducción
La mancha mongólica, también llamada melanocitosis dérmica congénita (MD), suele aparecer en el nacimiento o durante las primeras semanas de vida, aumenta en los 2 primeros años y después desaparece de modo gradual. A los 10 años la mayoría de estas manchas han remitido; si la mancha se mantiene en la edad adulta se denomina MD persistente. Su frecuencia, similar en ambos sexos, varía entre los distintos grupos raciales. El término 'mancha mongólica' se debe a su frecuencia elevada en las razas orientales, sobre todo en los mongoles, en quienes aparece en el 90% de los recién nacidos. Se observa en el 80% de los individuos de raza negra, en el 40% de los latinos y en menos del 10% de los caucasianos y judíos(1-3). La localización clásica es la región lumbosacra y las nalgas. Se conoce como MD atípica cuando se presenta fuera de esta región, como la espalda, los hombros, el cuero cabelludo y las extremidades.
Esta forma generalizada o atípica no es excepcional, ya que aparece en más del 3% de los niños asiáticos, indios americanos y negros(3-5).
Si bien la mayoría de las MD no tienen significado patológico, las formas atípicas, extensas o múltiples han sido descritas en asociación con algunas enfermedades lisosomales(6-8).
El objetivo de este trabajo es reportar dos casos de esta infrecuente asociación que se presentaron en nuestro hospital en un período de 12 meses.
Casos clínicos
Caso 1
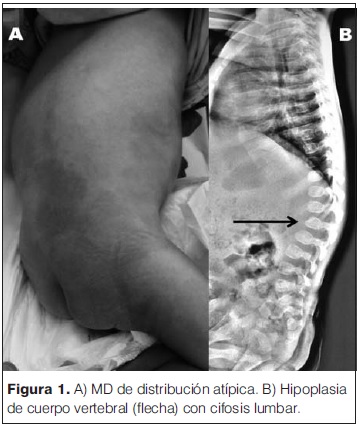
Lactante de 9 meses, sexo masculino, referido al hospital para evaluación por hipotonía, letargia y retraso global del desarrollo. Era un niño de raza caucasiana, producto de un primer embarazo sin complicaciones, de padres sanos adolescentes, no consanguíneos, sin antecedentes familiares de interés. Nacido a término por parto vaginal sin complicaciones, vigoroso, antropometría dentro de percentiles normales, sin patología perinatal a destacar. Sus primeros tres meses de vida presenta un desarrollo normal, luego de lo cual se objetiva un deterioro progresivo de las conductas adquiridas, presentando al momento de la consulta un severo retraso global del desarrollo. Al examen se destaca desnutrición severa, fascies toscas con cuello corto, puente nasal bajo, nariz ancha y filtrum largo, hipertrofia gingival, hepatomegalia leve, hidrocele bilateral, cifosis lumbar, no sostén cefálico, escasa fijación y seguimiento ocular, hipotonía global severa con reflejos osteotendinosos vivos y trastorno en la succión y deglución. El fondo de ojo mostró una retina pálida, no se evidenció mancha rojo cereza. A nivel cutáneo presenta una extensa mancha mongólica de color azulado y de distribución difusa a nivel del sector posterior de tronco y axilas (figura 1A). De la paraclínica se destaca la normalidad de: resonancia magnética de cráneo, perfil tiroideo, gasometría venosa, función renal y hepático, amoniemia, lactacidemia, aminoácidos en sangre, ácidos orgánicos en orina, uricemia, creatina quinasa en sangre y mucopolisacáridos en orina. La serología para citomegalovirus, rubeola y toxoplasmosis negativas. Los potenciales evocados visuales y auditivos presentaron una severa disfunción bilateral. En el frotis de sangre periférica se objetivaron linfocitos vacuolados. La radiografía de columna de perfil mostró una cifosis lumbar y vértebras con hipoplasia del sector ántero-superior del cuerpo vertebral (figura 1B). La presencia de oligosacáridos elevados en orina y la actividad deficiente de b-galactosidasa en sangre, presentando valores de 1,6 nmol/h/L (normal 30,4–128,4 nmol/h/L), confirmó el diagnóstico de gangliosidosis GM1. El paciente falleció a los 15 meses de vida.
Paciente 2
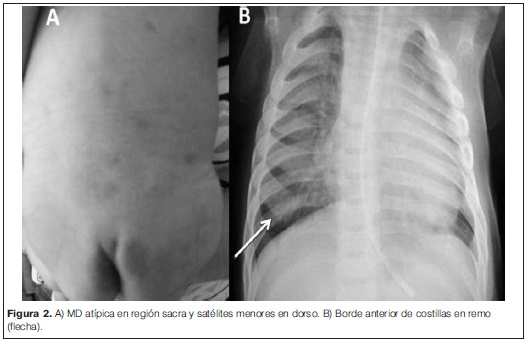
Lactante de 3 meses, sexo masculino, producto de una primera gestación sin complicaciones. Padres sanos, no consanguíneos. Sin patología del parto ni perinatal, datos antropométricos normales al nacer. Como antecedentes se destacan laringomalacia congénita, tres infecciones respiratorias que requirieron internación por insuficiencia respiratoria, retraso en el desarrollo motor, trastorno deglutorio y hernia inguinal derecha operada. Al examen se constata reactivo, sonrisa social, fija y sigue con la mirada, fascies tosca con retrognatia, narinas antevertidas, raíz nasal ancha y deprimida, sinofris, hepatomegalia, hipotonía global de predominio axial, no parética, con reflejos osteotendinosos vivos, clonus de pie izquierdo. A nivel de piel se evidencia extensa melanosis dérmica en dorso y múltiples satélites más pequeñas (figura 2A). La evaluación oftalmológica evidenció opacidades corneanas bilaterales. De la paraclínica se destaca: tomografía computada de cráneo, perfil tiroideo, gasometría venosa, función renal y hepática, creatina quinasa en sangre, amoniemia, lactacidemia y hemograma normales. La serología para citomegalovirus, rubeola y toxoplasmosis resultaron negativas. Los potenciales evocados visuales y auditivos mostraron una disfunción bilateral. La radiografía de tórax mostró un ensanchamiento del sector anterior de las costillas (costillas en remo) (figura 2B). Se objetivó un aumento de los mucopolisacáridos en orina. La deficiencia en la actividad enzimática de la a-L-iduronidasa en sangre, con valores de 0,6 mmol/l/h (normal mayor a 2,5 mmol/l/h), confirmó el diagnóstico de MPS tipo I.
Discusión
La asociación entre MD atípica y algunas enfermedades lisosomales fue descripta por primera vez por Weissbluth en 1981(9) y desde entonces fueron publicados varios casos incluyendo gangliosidosis GM 1, mucopolisacaridosis (MPS) I (enfermedad de Hurler), MPS II (enfermedad de Hunter), alfa manosidosis, Niemann-Pick, mucolipidosis II, MPS IV y enfermedad de Sandhoff(10-13). Las que se asocian más frecuentemente son la MPS I y la gangliosidosis GM1 (10).
Existen tres tipos de gangliosidosis GM1 de acuerdo a la edad de presentación clínica. La tipo 1, la forma más temprana y severa, como nuestro caso 1, se asocia a una MD atípica(14). Afecta a la infancia temprana, presentando mortalidad temprana cerca de los dos años de edad. Se trata de un proceso de base genética con mecanismo de herencia autosómico recesivo causado por el déficit de la enzima lisosomal b-galactosidasa, que da lugar a un aumento y depósito de gangliósido GM1 y otros esfingolípidos en distintos órganos provocando las características de la enfermedad. El fenotipo clínico es sugestivo, con fascies toscas, puente nasal aplanado, hipertrofia gingival, macroglosia, hipertricosis, hepatoesplenomegalia, retraso psicomotor, hipotonía y retraso del desarrollo, convulsiones y la presencia de mancha rojo cereza en la mácula. El estudio radiológico evidencia elementos de displasia esquelética como deformación de los cuerpos vertebrales y de la silla turca, anomalías de los huesos largos y de la pelvis. Como ya dijimos, los niños raramente sobreviven más allá de los 3 años(5,10,15).
La MPS tipo I es la forma más frecuente de MPS. Es producida por un déficit de la enzima a-L-iduronidasa produciendo la acumulación en los tejidos y el aumento en la excreción urinaria de dermatán y heparánsulfato. Es también una enfermedad lisosomal y comparte muchos rasgos clínicos con la gangliosidosis GM1 tipo 1 o infantil. Alrededor de los 6 meses de edad aparecen opacidades corneales, visceromegalias, disostosis múltiple y rasgos faciales toscos. Provoca retraso mental, infecciones respiratorias a repetición y cardiopatía mortal alrededor de los 10 años de edad(10). La RNM de cráneo revela alteración en la señal de la sustancia blanca, aumento del espacio subaracnoideo, dilatación de los espacios perivasculares y ventriculares.
El diagnóstico de estas enfermedades se hace a través del aumento en la excreción urinaria de heparán y dermatán sulfato para MPS I y de oligosacáridos para la gangliosidosis GM 1. La confirmación definitiva se logra mediante la demostración de las deficiencias enzimáticas en leucocitos o en sangre para b-galactosidasa y a-L-iduronaidasa en GM 1 y MPS I respectivamente. Un diagnóstico temprean en el caso de MPS I permite realizar un tratamiento específico como es la terapia de remplazo enzimático con el consiguiente beneficio sobre todo en la sintomatología pulmonar y la resolución de la hepatoesplenomegalia(16).
No se sabe cuál es la causa de la asociación entre las enfermedades lisosomales y la MD atípica. Se plantea que los gangliósidos, que son glicoesfingolípidos que se encuentran en las membranas plasmáticas de las células, participan en varios procesos de interacción extracelular(17). La acumulación de estos metabolitos que se produce en la GM1 se vincularía estrechamente a un receptor del factor de crecimiento neuronal (FCN) tipo tirosinquinasa (Trk). Dicha unión estimularía al receptor, provocando manifestaciones neurológicas. En cultivos celulares se ha visto que estos receptores también se encuentran en los melanocitos y que al ser activados funcionan como un factor quimiotáctico. Una hipótesis es que la melanocitosis dérmica en niños con estas enfermedades lisosomales sea el resultado de la acumulación de gangliosidos (en la gangliosidosis GM1) y heparán sulfato (en la MPS I), los cuales mediante la estimulación de FCN a través de Trk, detendrían la migración transdérmica de los melanocitos(11).
En conclusión la presencia de una extensa melanocitosis dérmica y un contexto fenotípico característico, puede llevarnos a la sospecha temprana de una enfermedad lisosomal, permitiendo un apropiado asesoramiento genético y orientación anticipada sobre el curso típico evolutivo de estas enfermedades e inicio, en los casos como en la MPS 1, de un tratamiento específico temprano: terapia de remplazo enzimático.
Referencias bibliográficas
1. Egemen A, Ikizoðlu T, Ergör S, Mete G, Yilmaz O. Frequency and characteristics of mongolian spots among Turkish children in Aegean region. Turk J Pediatr 2006; 48(3):232-6.
2. Avilés JA, Hernanz JM, De la Cueva P. Mancha mongólica. Acta Pediatr Esp 2004; 62(1):60-1.
3. Valdés F, Ginarte M, Toribio J. Melanocitosis dérmicas. Actas Dermosifiliogr 2001; 92(9):379-88.
4. Torrelo A, Zambrano A, Happle R. Large aberrant Mongolian spots coexisting with cutis marmorata telangiectatica congenita (phacomatosis pigmentovascularis type V or phacomatosis cesiomarmorata). J Eur Acad Dermatol Venereol 2006; 20(3):308-10.
5. Torrelo A, Solana L, Mediero I, Ruiz-Falco ML, García-Peñas JJ, Zambrano A. Mancha mongólica generalizada en un niño con gangliosidosis GM1 tipo 1. Actas Dermosifiliogr 2000; 91(1):29-38.
6. Vedak P, Sells R, De Souza A, Hoang MP, Kroshinsky D. Extensive and Progressing Congenital Dermal Melanocytosis Leading to Diagnosis of GM1 Gangliosidosis. Pediatr Dermatol 2015; 32(6):e294-5.
7. Armstrong-Javors A, Chu C. Child neurology: Exaggerated dermal melanocytosis in a hypotonic infant: a harbinger of GM1 gangliosidosis. Neurology 2014; 83(17):e166-8.
8. Ashrafi MR, Tavasoli A, Shiva S, Parvaneh N, Tamizifar B. Diffuse dermal melanocytosis in two patients with Sandhoff disease and mucopolysaccharidosis VI. Int J Dermatol 2014; 53(6):736-8.
9. Weissbluth M, Esterly N, Caro W. Report of an infant with GM1 gangliosidosis type I and extensive and unusual mongolian spots. Br J Dermatol 1981; 104(2):195-200.
10. Ashrafi MR, Shabanian R, Mohammadi M, Kavusi S. Extensive Mongolian spots: a clinical sign merits special attention. Pediatr Neurol 2006; 34(2):143-5.
11. Hanson M, Lupski JR, Hicks J, Metry D. Association of dermal melanocytosis with lysosomal storage disease: clinical features and hypotheses regarding pathogenesis. Arch Dermatol 2003; 139(7):916-20.
12. Lee S, Kim D, Lee G, Whang K, Lee JS, Park Y. An unusual case of congenital dermal melanocytosis. Ann Dermatol 2010; 22(4):460-2.
13. Su F, Li F, Jin H. Extensive Mongolian spots in a child with mucolipidosis II. Int J Dermatol 2010; 49(4):438-40.
14. Selsor L, Lesher JL. Hyperpigmented macules and patches in a patient with GM1 type 1 gangliosidosis. J Am Acad Dermatol 1989; 20(5 Pt 2):878-82.
15. Yuste M, Román C, González A, González P, Aramendi T. Mancha mongólica generalizada. Actas Dermosifiliogr 1999; 90(7):373-7.
16. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab 2014; 111(2):63-72.
17. Yanagisawa M, Liour S, Yu R. Involvement of gangliosides in proliferation of immortalized neural progenitor cells. J Neurochem 2004; 91(4):804-12.
Correspondencia: Dr. Conrado Medici.
Correo electrónico: conrado.medici@gmail.com

