










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versão On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.80 no.3 Montevideo set. 2009
ARTÍCULO ORIGINAL
Arch Pediatr Urug 2009; 80(3)
Encefalomielitis difusa aguda en la edad pediátrica
Presentación de una casuística nacional y de las propuestas de definiciones consensuadas para esclerosis múltiple y trastornos relacionados en pediatría del consenso de expertos del International Pediatric MS Study Group
Dres. Gabriela Capote 1, Alfredo Cerisola 2, Gabriel González 3, Sully López 4, Cristina Scavone 5
1. Asistente de la Cátedra de Neuropediatría.
2. Profesor Adjunto de la Cátedra de Neuropediatría.
3. Profesor Agregado de la Cátedra de Neuropediatría.
4. Neuropediatra.
5. Profesora Directora de la Cátedra de Neuropediatría.
Cátedra de Neuropediatría, Facultad de Medicina, Universidad de la República. Servicio de Neuropediatría, Centro Hospitalario Pereira Rossell. Bvar. Artigas 1550. Montevideo. Uruguay.
Fecha recibido: 28 de setiembre de 2009.
Fecha aprobado: 1 de diciembre de 2009.
Resumen
La encefalomielitis diseminada aguda (EMDA) es una enfermedad inflamatoria aguda del sistema nervioso central, inmunomediada, que es más frecuente en la edad pediátrica. Su diagnóstico se basa en la clínica, en la confirmación del proceso desmielinizante agudo a través de la resonancia magnética y en la exclusión de otras posibles etiologías de acuerdo con los criterios propuestos en el consenso de expertos del “International Pediatric MS Study Group”. La evolución de los conocimientos y los nuevos criterios diagnósticos justifican la revisión del tema. Se realizó un estudio descriptivo y retrospectivo sobre la base de la revisión de las historias clínicas de una serie consecutiva de 12 pacientes menores de 15 años que ingresaron al Hospital Pediátrico del Centro Hospitalario Pereira Rossell con diagnóstico de EMDA desde el 1 de enero del 2000 al 31 de diciembre del 2007 y su seguimiento posterior en la Cátedra de Neuropediatría de la Facultad de Medicina y el Servicio de Neuropediatría de dicho hospital. Si bien se presentaron con una encefalopatía aguda severa, la evolución fue buena en la mayoría de los pacientes, aunque 2 pacientes presentaron secuelas motoras y uno epilepsia secundaria y compromiso cognitivo. En 11 pacientes la enfermedad tuvo un curso monofásico. La importancia del seguimiento clínico e imagenológico radica en que existen formas menos frecuentes con recaídas o recurrencias. También debe considerarse la posibilidad de que un primer evento desmielinizante, indistinguible de una EMDA, constituya el debut de una esclerosis múltiple, como ocurrió en un caso de esta serie.
Palabras clave:
ENCEFALOMIELITIS AGUDA DISEMINADA
ESCLEROSIS MÚLTIPLE
IMAGEN POR RESONANCIA MAGNÉTICA
Summary
Acute disseminated encephalomyelitis (ADEM) is an acute inflammatory disease of the central nervous system, immune mediated, which is more common in childhood. Its diagnosis is based on clinical confirmation of acute demyelinating process through the MRI and the exclusion of other diagnoses according to the criteria proposed in the consensus of experts of the International Pediatric MS Study Group. The evolution of knowledge and new diagnostic criteria justify a revision of the item. We performed a retrospective descriptive study based on review of medical records of a consecutive series of 12 patients under age 15 admitted to the Pediatric Hospital Pereira Rossell Hospital Center with diagnosis of ADEM from 1 January 2000 to December 31, 2007 and its subsequent monitoring at the Chair of Pediatric Neurology, Faculty of Medicine and the Pediatric Neurology Service of the hospital. While presented with severe acute encephalopathy, the outcome was good in most patients, although two patients had motor sequelae and one secondary epilepsy and cognitive impairment. In 11 patients the disease had a monophasic course. The importance of clinical and imaging follow-up is that there are ways less frequent relapse or recurrence. It should also be considered a first demyelinating event, indistinguishable from ADEM, constitutes the debut of multiple sclerosis, as in a case of this series.
Key words:
ENCEPHALOMYELITIS , ACUTE DISSEMINATED
MULTIPLE SCLEROSIS
MAGNETIC RESONANCE IMAGING
Introducción
La encefalomielitis diseminada aguda (EMDA) es una enfermedad inflamatoria aguda del sistema nervioso central (SNC), inmunomediada, habitualmente precedida por una infección inespecífica o más raramente vacunación, que involucra predominantemente la sustancia blanca (SB) supra e infratentorial y medular y con menor frecuencia la sustancia gris (SG) cortical y profunda (1).
Si bien puede ocurrir a cualquier edad, es más común en la infancia, con un discreto predominio en varones y con una distribución estacional, predominando en invierno y primavera (1-3). Considerada una enfermedad poco frecuente en la literatura clásica, su incidencia se ha duplicado en los últimos años debido probablemente al desarrollo y uso más extendido de la resonancia magnética (RM), permitiendo una adecuada identificación de esta patología (2,4-6). La incidencia real es difícil de establecer, considerándose actualmente de 0,4/100.000 habitantes menores de 20 años por año (1,3).
Clínicamente se caracteriza por un inicio agudo o subagudo, polisintomático, poco específico y muy variable, con síntomas neurológicos dominantes que deben incluir alteración de la conciencia, de curso habitualmente monofásico, pudiendo presentar recurrencias (1,4,7,8).
El diagnóstico se basa en la clínica, en la confirmación del proceso desmielinizante agudo a través de la RM y la exclusión de otras posibles etiologías, sobre todo infecciosas (9-15), de acuerdo con los criterios propuestos en el consenso de expertos del International Pediatric MS Study Group para EMDA monofásica, EMDA recurrente y EMDA multifásica (1,7) (Anexo 1).
La evolución es a la mejoría tras el tratamiento en hasta el 75% de los pacientes, con una mortalidad actual inferior al 10% (5,16-21).
Si bien existe a nivel nacional una comunicación previa no publicada sobre esta patología, la evolución de los conocimientos relativos a la misma y los nuevos criterios diagnósticos establecidos, justifican la revisión del tema y el análisis de la casuística planteada (22).
Objetivo
Describir la forma de presentación clínica, los hallazgos licuorales, neuroimagenológicos y electrofisiológicos, los tratamientos realizados y la evolución a largo plazo, de una serie de pacientes con diagnóstico de EMDA que ingresaron al Centro Hospitalario Pereira Rossell (CHPR) entre los años 2000 y 2007.
Pacientes y método
Estudio descriptivo y retrospectivo sobre la base de la revisión de las historias clínicas de una serie consecutiva de pacientes menores de 15 años que ingresaron al Hospital Pediátrico del CHPR con diagnóstico de EMDA desde el 1 de enero del 2000 al 31 de diciembre del 2007 y su seguimiento posterior en la Cátedra de Neuropediatría de la Facultad de Medicina y el Servicio de Neuropediatría de dicho hospital.
Criterios de inclusión
Se incluyeron aquellos pacientes que cumplían en su debut con los criterios propuestos para el diagnóstico de EMDA según el consenso elaborado por el International Pediatric MS Study Group (7), identificados en el análisis de las diferentes bases de datos existentes en el CHPR.
Registro de datos
Se elaboró un formulario especial para esta investigación con el fin de registrar la información de las historias clínicas. Se registraron todas las hipótesis diagnósticas planteadas y los estudios realizados para descartarlas, al igual que datos relevantes del tratamiento y del pronóstico inmediato.
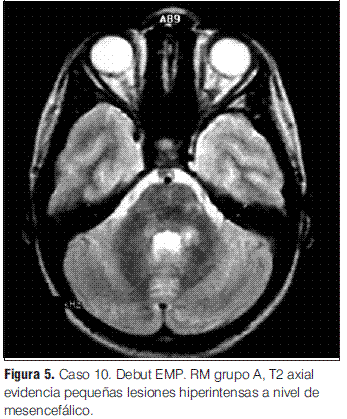
Las variables analizadas fueron: sexo, edad de inicio, infecciones o vacunaciones precedentes, síntomas y signos de presentación, hallazgos en la neuroimagen (RM) (clasificada de acuerdo a los criterios de Tenembaun y colaboradores [Anexo 2] (4,19), respuesta terapéutica y evolución de la enfermedad, especificándose número de controles clínicos y de RM, tiempo transcurrido entre el evento inicial, el control y los hallazgos en los mismos.
Para establecer la magnitud del compromiso neurológico se utilizó la Escala Ampliada del Estado de Discapacidad de Kurtzke (EDSS) [Anexo 3] (23) al egreso y en cada control, infiriéndose de la historia clínica si no estaba específicamente establecido su valor.
Análisis estadístico
Las variables se expresaron en frecuencias absolutas y relativas. Las variables cuantitativas se expresaron mediante medidas de tendencia central (media y mediana) y medidas de dispersión (rango).
Resultados
En el período comprendido entre el 1 de enero del 2000 y el 31 de diciembre del 2007 ingresaron al CHPR 19 pacientes en los cuales se planteó el diagnóstico de probable EMDA. Del total de pacientes, 12 cumplieron con los criterios de inclusión (frecuencia relativa [FR] 0,63). De los siete que se excluyeron a partir de la revisión de las historias clínicas surge que dos casos se presentaron como clínica polisintomática pero sin encefalopatía (FR 0,10), dos como eventos desmielinizantes aislados (FR 0,10) (un paciente con síndrome de tronco encefálico y otro con ataxia cerebelosa), uno correspondió a una cerebelitis varicelosa intrainfecciosa (FR 0,05) y dos a encefalitis virales no herpéticas (FR 0,10).
La distribución anual de casos mostró un paciente en 2000, dos en 2001, un paciente en 2002, dos en 2003, dos en 2005, tres en 2006 y uno en 2007. Considerados en conjunto, tres casos se presentaron en verano, cuatro en otoño, cuatro en primavera y uno en invierno.
Se incluyeron cuatro varones y ocho niñas, con una edad media de 7,4 años y una mediana de 7 años (rango de edades: 2 a 13 años). Los 12 pacientes requirieron en total 492 días de hospitalización, con una media de 41 días y una mediana de 30 días (rango: 11-180 días). De los 12 pacientes 10 (FR 0,83) requirieron su ingreso en una unidad de cuidados intensivos pediátricos (UCIP), requiriendo siete de ellos asistencia ventilatoria mecánica (AVM) durante una media de 6 días (rango de 2 a 16 días).
El principal diagnóstico diferencial al ingreso fue la infección del SNC (planteada en 10 de los 12 pacientes), realizándose tratamiento empírico antibiótico y antiviral hasta haber descartado la misma. Otros diagnósticos planteados fueron accidente cerebrovascular, vasculitis primarias o secundarias del SNC e intoxicaciones agudas.
Las características clínicas, epidemiológicas y evolutivas de la población se resumen en las tablas 1 y 2.

En todos los pacientes se realizó en LCR (con el objetivo de descartar otros diagnósticos) el estudio citoquímico, bacteriológico y virológico –a través de reacción en cadena de la polimerasa (PCR) para el estudio de los virus de la familia herpes–, proteinograma electroforético, Index IgG y bandas oligoclonales (BOC); dos pacientes (FR 0,16) presentaron alteraciones caracterizadas en un caso por hiperproteinorraquia de 216 mg/dl, pleocitosis linfocitaria (86 linfocitos/mm3) y aumento del Index IgG, y en el segundo caso por hiperproteinorraquia de 186 mg/dl, sin otras alteraciones. En ningún paciente se hallaron BOC. Los estudios bacteriológicos y virológicos fueron negativos.
Se efectuaron electroencefalogramas (EEG) en 10 pacientes, mostrando en nueve de ellos enlentecimiento del ritmo de fondo y en ocho actividad paroxística de topografía variable.
Los potenciales evocados visuales (PEV) y los potenciales evocados auditivos de tronco (PEAT) se solicitaron en tres pacientes, mostrando uno de ellos disfunción bilateral en el PEV.
Se efectuó estudio de la velocidad de conducción nerviosa (VC) y electromiograma (EMG) en un paciente con clínica compatible con afectación del sistema nervioso periférico (SNP), evidenciándose afectación de VC motora y sensitiva de miembros inferiores (MMII), con denervación severa en el territorio de las raíces L3-S1.
Se realizaron RM en la primera semana de evolución de la enfermedad en todos los pacientes, revelando en todos los casos alteraciones características (tabla 3).
Todos recibieron tratamiento con corticoides inicialmente con tratamientos en altas dosis por vía intravenosa (11 pacientes recibieron bolos de metilprednisolona, la mayoría de 3 a 5 días, y uno recibió dexametasona). Los esquemas de tratamiento fueron muy diversos. La duración del tratamiento corticoideo ha ido disminuyendo en los últimos años, desde períodos de un año en los primeros casos de esta serie, hasta la conducta actual de tratamientos menos prolongados (aproximadamente 2 meses). Un paciente recibió IgG y ninguno recibió plasmaféresis.
La situación clínica al alta evaluada a través de la EDSS, en relación a los hallazgos en la RM inicial, se resume en la tabla 3.
En 11 pacientes (FR 0,92) se pudo controlar la evolución, con una media de tiempo de seguimiento clínico de 3 años (rango de 12 meses a 6 años): 8/11 (FR 0,73) no presentaban secuelas al momento de último control, 2 (FR 0,18) presentaban secuelas motoras y 1 paciente (FR 0,09) epilepsia secundaria y nivel cognitivo globalmente descendido (tabla 3).
No observamos casos de corticodependencia.
En cuanto a las RM de control, siete se normalizaron entre el año y los 3 años 7 meses; las cuatro en las que persistieron lesiones correspondieron tres de ellas a los pacientes con secuelas (tanto motoras como epilepsia secundaria y nivel cognitivo descendido) y una a un paciente que evolucionó a la esclerosis múltiple pediátrica (EMP) (tabla 3).
Discusión
El CHPR constituye el único hospital pediátrico del MSP en el Uruguay y es el centro de referencia nacional para la atención pediátrica del MSP. La mayoría de los pacientes del interior del país con patologías neurológicas a quienes se les solicitan estudios de complejidad, como la RM, son derivados al CHPR, donde atiende la Cátedra de Neuropediatría de la Facultad de Medicina y el Servicio de Neuropediatría del MSP.
Del total de aproximadamente 775.606 niños menores de 15 años residentes en el Uruguay, el 61% (585.000) se asistían en el Ministerio de Salud Pública (MSP) hasta el 31 de diciembre de 2007 (24,25). En base a la incidencia calculada de EMDA en menores de 20 años de 0,4 cada 100.000 habitantes a partir de estudios poblacionales en otros países (1,2,10), se realizó una estimación de la incidencia teórica esperada de EMDA en la población de niños asistidos en el MSP. Los 12 pacientes de la serie representan un promedio anual de 1,5 pacientes, con una incidencia calculada de 0,25 cada 100.000 habitantes menores de 15 años atendidos en el MSP por año, valor inferior a la frecuencia teórica estimada de 2,3 pacientes por cada 100.000 habitantes menores de 20 años por año o al valor absoluto estimado de 18,4 pacientes para el período de ocho años en dicha población.
No se incluyeron a los pacientes con edades comprendidas entre los 15 y los 20 años dado que en nuestro país, a partir de los 15 años de edad, la población usuaria del MSP se asiste en centros hospitalarios destinados a pacientes adultos. No hemos encontrado en la literatura nacional revisiones de esta patología que nos permitan comparar estos hallazgos.
A partir de estas consideraciones podemos suponer que la EMDA es una patología subdiagnosticada en nuestro país. Un número importante de pacientes con diagnóstico de “encefalopatía aguda” de etiología no definida, podrían no llegar al CHPR, no realizándose RM por no disponer de la misma en los departamentos del interior del país.
En cuanto a las características clínicas y epidemiológicas de la serie, el rango de edades y la edad media de la población, al igual el antecedente de infección dentro del mes previo al debut clínico es similar a otras series publicadas (2-4,21,26). A diferencia de éstas, en la nuestra predominó el sexo femenino, la distribución estacional mostró que más de la mitad de los casos se presentaron en verano y otoño, y no observamos casos vinculados a vacunaciones. En las series mencionadas se describe un discreto predominio en varones y, en relación a la distribución estacional, un predominio en invierno y primavera. Estas diferencias podrían deberse al pequeño tamaño de la muestra.
Con respecto a la presentación clínica, tres de cada cuatro pacientes debutaron con síntomas y signos neurológicos de gravedad tal que requirió su ingreso a UCIP, con ventilación asistida en aproximadamente el 90% de éstos. La estadía hospitalaria promedio fue prolongada.
El principal diagnóstico diferencial planteado fue la infección del SNC y, al igual que en todas las series publicadas, fue la primera etiología a descartar a través del estudio del LCR, realizándose tratamiento empírico antibiótico y antiviral hasta haber descartado la misma (1).
La situación clínica evaluada a través de la EDSS evidenció importante discapacidad al momento del alta hospitalaria, pero con franca mejoría clínica en la evolución, con 64% de los pacientes sin secuelas entre 1 año y 3 años 7 meses luego del evento (tabla 3). No se registraron fallecimientos en este período; estas observaciones son coincidentes con la mayoría de las series publicadas (2,4,27).
En cuanto a los exámenes solicitados, la baja frecuencia de hallazgos inespecíficos sugerentes de inflamación a nivel del LCR es acorde con otras series publicadas (1,2,4). El estudio del LCR está orientado fundamentalmente a descartar la infección del SNC, principal diagnóstico diferencial al ingreso.
La mayoría de los EEG mostraron enlentecimiento de la actividad de fondo y descargas epileptiformes, hallazgos inespecíficos en relación a esta patología, como está descrito en otras series (2,4). Los PEV mostraron alteraciones en el paciente que evolucionó a la EMP, lo cual apoyaría la utilidad de los estudios electrofisiológicos en la detección de lesiones desmielinizantes asintomáticas frecuentes en niños con EMP (2,26,27). Los PEAT fueron normales en los dos pacientes evaluados con esta técnica.
En el paciente con afectación concomitante del SNC y SNP el estudio electrofisiológico de los cuatro miembros (VC y EMG) fue fundamental para definir el compromiso periférico; este estudio no fue solicitado de rutina sino frente a la sospecha clínica; la concomitancia del compromiso del SNC y del SNP ha sido comunicada en diferentes estudios; futuras investigaciones buscando en forma sistemática esta asociación serán necesarias para aclarar la incidencia, los factores de riesgo, las posibles etiologías y el manejo terapéutico más adecuado, al igual que el pronóstico en estos casos (28,29).
Si bien todas las series publicadas describen una buena respuesta terapéutica a los corticoides en dosis altas, el tiempo de tratamiento a dosis plenas y el de descenso está en permanente revisión. No hay ensayos clínicos controlados que permitan asegurar el papel decisivo de los corticoides (habiendo varias publicaciones que informan sobre la remisión espontánea de la enfermedad), ni los tiempos ideales de duración del tratamiento (2,4,5).
El estudio prospectivo de Tenembaun y colaboradores con una amplia cohorte concluye que los corticoides a altas dosis acortan y mejoran la recuperación neurológica en la mayoría de los pacientes, haciéndose evidente la mejoría clínica en horas desde el inicio del tratamiento; no se observó beneficio con dosis bajas de corticoides ni con tratamientos prolongados (4,9,11).
Se plantea el uso de IgG como tratamiento de segunda línea frente a una mala respuesta al tratamiento con corticoides o como coadyuvante desde el inicio en casos graves; también la plasmaférisis podría ser una opción si fracasaran los tratamientos anteriores (18,20,30,31).
Podemos decir, coincidiendo con otros autores de estudios con casuísticas publicadas, que si bien la EMDA es una enfermedad infrecuente se presenta habitualmente como una enfermedad grave, que insume importantes gastos al sistema de salud y determina una morbilidad severa en etapas iniciales, pero que en general presenta una buena evolución clínica en cuanto al pronóstico vital alejado. Las secuelas se producen en alrededor del 20% de los casos, pero el porcentaje de discapacidad grave en la evolución (puntaje de cuatro o más según la escala de Kurtzke) es bajo (2,4,9,21).
En general las secuelas son motoras, pero actualmente se describen alteraciones emocionales, conductuales y cognitivas significativas (32). En nuestra serie de un total de cuatro pacientes con secuelas (FR 0,30), tres presentaron secuelas motoras, uno de ellos con secuelas esfinterianas asociadas y un paciente presentó retardo mental, trastornos conductuales y epilepsia coincidiendo con una mayor afectación, tanto clínica (crisis convulsivas) como en la RM de la SG durante el evento agudo (tabla 3).
La persistencia en la evolución de lesiones en la RM se correlaciona con la presencia de secuelas clínicas, pero no encontramos en la literatura ni en nuestra serie una relación significativa entre el grupo de la RM inicial y la evolución posterior en cuanto la posibilidad de secuelas y grado de discapacidad; sí se relaciona el compromiso de la SG con la posibilidad de secuelas cognitivas y epilepsia secundaria, pero esto requiere de estudios más amplios y seguimientos prolongados. Serán necesarias futuras investigaciones para definir las causas o bases etiopatogénicas de las diferentes características clínicas y evolutivas de los pacientes.
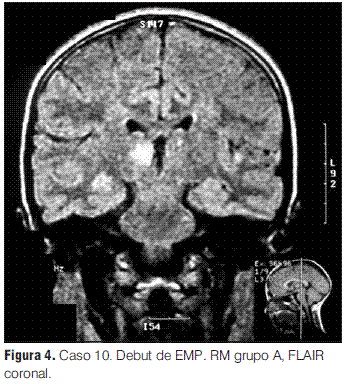
En nuestra serie no se presentaron recaídas ni recurrencias, con una media de tiempo de seguimiento de 3 años y 1 mes. Sin embargo, de los 12 pacientes con diagnóstico inicial de EMDA incluidos en este estudio, uno evolucionó a una esclerosis múltiple pediátrica (EMP), de acuerdo con los criterios propuestos por el consenso de expertos del “International Pediatric MS Study Group”, representando el 0,09 de la población en la que se pudo realizar seguimiento; la proporción de pacientes que tienen esta evolución varía en las diferentes series entre el 9% y el 40% (2,3); la presentación inicial de esta paciente con clínica polisintomática y con compromiso de conciencia (sintomatología EMDA-like) es una forma frecuente de debut de la EMP, lo cual subraya la importancia del seguimiento clínico e imagenológico sistematizado y prolongado para poder llegar a un diagnóstico definitivo [comunicación personal Tenembaun y colaboradores (33)]. Destacamos la afectación de los PEV sin repercusión clínica en este paciente, como signo de lesiones desmielinizantes silentes (4,2,17,27,28,34,35).
A partir de los hallazgos en nuestra serie y de la revisión de la literatura internacional podemos decir que si bien la evolución, a mediano y largo plazo, es buena en la mayoría de los pacientes, debe enfatizarse la importancia del seguimiento evolutivo clínico e imagenológico, ya que si bien la mayoría de las EMDA se comportan como enfermedades monofásicas existen formas menos frecuentes con recaídas y recurrencias y el riesgo de que un primer evento desmielinizante con características clínicas indistinguibles de una EMDA constituya en realidad el debut de una EMP está siempre presente.
La investigación sistemática del compromiso del SNP a través de estudios electrofisiológicos (VC y EMG) está aún en discusión, desconociéndose la frecuencia real de esta concomitancia.
El seguimiento debe incluir la evaluación neuropsicológica, ya que si bien las secuelas motoras son las más frecuentes también pueden presentarse secuelas cognitivas.
Referencias bibliográficas
1. Tenembaum S, Chitnis T, Ness J, Hahn JS; International Pediatric MS Study Group. Acute disseminated encephalomyelitis. Neurology 2007 Apr 17; 68(16 Suppl 2): S23-36.
2. Erazo Torricelli R. Encefalomielitis aguda diseminada en la niñez. Rev Neurol 2006; 42 (supl 3): S75-S82.
3. Leake JAD, Albani S, Kao AS, Senac MO, Billman GF, Nespeca MD, et al. Acute Disseminated Encephalomyelitis in childhood: Epidemiologic, clinical and laboratory features. Pediatr Infect Dis J 2004; 23: 756-63.
4. Tenembaun S. Encefalomielitis diseminada aguda. Estudio prospectivo de una cohorte pediátrica. Med Infant 2005; 12: 180-91.
5. Menge T, Hemmer B, Nessler S, Wiendl H, Neuhause O, Hartung HP, et al. Acute Disseminated Encephalomyelitis: an up to date Arch Neurol 2005; 62: 1673-80.
6. Neuteboom RF, Boon M, Catsman Berrevoets CE, Vles JS, Gooskens RH, Stroink H, et al. Prognostic factors after a first attack of inflammatory CNS demyelination in children. Neurology 2008; 71:967-973.
7. Krupp LB, Banwell B, Tenembaum S; International Pediatric MS Study Group. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007 Apr 17; 68(16 Suppl 2): S7-12.
8. Pohl D. Epidemiology, immunopathogenesis and management of pediatric central nervous system inflammatory demyelinating conditions. Curr Opin Neurol 2008; 21(3): 366-72.
9. Gómez-Gosálves FA, Smeyers P, Menor F, Morant A, Carbonell J, Mulas F. Encefalomielitis aguda diseminada en la infancia. Estudio retrospectivo de siete pacientes. Rev Neurol 2000; 30(4): 304-10.
10. Guitet M, Campistol J, Cambra FJ. EMDA en la infancia, presentación de 10 casos. Rev Neurol 2001; 32 (5): 409-13.
11. Martinez Anton A, Ramos Amador JT, Rojo Conejo P, Gómez Sánchez E, Salinas Sanz JA. Respuesta espectacular de una Encefalomielitis Aguda Diseminada tras altas dosis de esteroides. An Pediatr (Barc) 2005; 63(5): 457-68.
12. Krishna Murthy SN, Faden HS, Cohen ME, Bakshi R. Acute disseminated encephalomyelitis in children [en línea] Pediatrics 2002; 110 (2): e21. Obtenido de: http://www.pediatrics.org [consulta: 31 dic. 2007].
14. Hynson JL, Kornberg AJ, Coleman LT, Shield L, Harvey AS, Kean MJ. Clinical and Neuroradiologic features of ADE in children. Neurology 2001; 56: 1308-12.
15. López-Pisón J, García-Bodega O, Díaz-Suárez M, Bajo-Delgado AF, Cabrerizo de Diago R, Peña-Segura JR. Inflamación diseminada episódica del SNC en niños. Revisión casuística de un período de 13 años. Rev Neurol 2004; 38(5): 405-10.
16. Peña JA, Montiel-Nava C, Hernández F, Medrano E, Valbuena O, Cardozo J. Encefalomielitis aguda diseminada en niños. Rev Neurol 2002; 34(2): 163-8.
17. Mikaeloff Y, Suissa S, Vallée L, Lubetzki C, Ponsot G, Confavreux C, et al. First Episode of Acute CNS Inflammatory Demyelination in Childhood: prognosis factors for ME and Disability. J Pediatr 2004; 144: 246-52.
18. Shahar E, Andraus J, Savitzki D, Pilar G, Zelnik N. Outcome of severe encephalomyelitis in children: effect of high-dose methylprednisolone and inmunoglobulins. J Child Neurol 2002; 17(11): 810-4.
19. Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis. A long–term follow-up study of 84 pediatric patients. Neurology 2002; 59: 1224-31.
20. Gómez Sánches E, Mateos Beato F, Sánchez Díaz JI, Simón de las Heras R, Ballesteros Díaz Y. Encefalomielitis aguda. Experiencia de un hospital terciario español. An Pediatr (Barc) 2005; 63(3): 203-11.
21. Gupte G, Stonehause M, Wassmer E, Coad NAG, Whitehouse WP. Acute disseminated encephalomyelitis: a review of 18 cases in childhood. J. Pediatr Child Health 2003; 39: 336-42.
22. Rossi M. EMDA, presentación de 5 pacientes pediátricos. [monografía postgrado neuropediatría]. Montevideo: UDELAR, Fac. Medicina, 2002.
23. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444-52.
24. Uruguay. Instituto Nacional de Estadísticas. Censo fase 1 (2004). Montevideo: INE, 2004. Obtenido de: http://www.ine.gub.uy [consulta: 31 dic. 2007].
25. Uruguay. Instituto Nacional de Estadísticas. Encuesta continua de hogares (2005). Montevideo : INE, 2005. Obtenido de: http://www.ine.gub.uy. [consulta: 31 dic.2007].
26. Brass SD, Caramanos Z, Santos C, Dilenge ME, Lapierre Y, Rosenblatt B. MS vs. ADEM in childhood. Pediatr Neurol 2003; 29: 227-31.
27. Dale RC, De Sousa C, Chong WK, Cox TSC, Harding B, Neville BGR. Acute disseminated encephalomyelitis, multiphase disseminated encephalomyelitis and multiple sclerosis in children. Brain 2000; 123: 2407-22.
28. Adamovic T, Riou EM, Bernard G, Banase M, Décarie J-C, Poulin C, et al. Acute combined central and peripheral nervous system demyelination in children. Pediatr Neurol 2008; 39: 307-16.
29. Bernard G, Riou EM, Rosenblatt B, Dilenge ME, Poulin C. Simultaneous Guillain-Barré Syndrome and Acute Disseminated Encephalomyelitis in Pediatric Population. J Child Neurol 2008; 23(7): 752-7.
30. Khurana DS, Melvin JJ, Kothare SV, Valencia I, Hardison HH, Yum S, et al. Acute Disseminated Encephalomyelitis in children: Discordant Neurologic and Neuroimaging Abnormalities and Response to Plasmapherisis. Pediatrics 2005; 116(2): 431-6
31. Meca-Lallana JE, Rodríguez-Hilario H, Martinez-Vidal S, Saura-Luján I. Plasmaférisis: su utilidad en la esclerosis múltiple y otros procesos desmielinizantes del sistema nervioso central. Estudio observacional. Rev Neurol 2003; 37 (10): 917-926
32. Jacobs RK, Anderson VA, Neale JL, Shield LK, Kornberg AJ. Neuropsychological outcome after acute disseminate encephalomyelitis: impact of age at illness onset. Pediatr Neurol 2004; 31: 191-7.
33. Tenembaun S, Moreira M, Callegaro D, Lana-Peixoto M, Peña J, D’Onghia A, et al. Clinical and demographic features of Pediatric Multiple Sclerosis. Preliminary data from a Latin America Multinational Collaborative Study Group. Poster presentado en el Congreso Mundial de EM. Montreal, Canadá. 17-20 set. 2008.
34. Dale RC, Pillai SC. Early relapse risk after a first CNS inflammatory demyelination episode: examining international consensus definitions. Dev Med Child Neurol 2007; 49(12): 887-93.
35. Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol 2007; 6(10): 887-902.
Correspondencia: Dr. Alfredo Cerisola
Correo elctrónico: alfredocerisola@gmail.com
Propuesta de definiciones consensuadas
para EM y trastornos relacionados en pediatría (7)
EMDA
Primer evento clínico de causa presumiblemente inflamatoria o desmielinizante, de inicio agudo o subagudo, que afecta áreas multifocales del SNC. La presentación clínica debe ser polisintomática y debe incluir:
alteración de la conciencia, por ejemplo letargia, coma.
2. El evento debe ser seguido de mejoría, tanto clínica como en la RNM, aunque pueden quedar secuelas.
En casos poco frecuentes la RNM muestra una lesión grande, mayor de 1-2 cm, única, que afecta la S
RM de médula puede mostrar lesión/lesiones intramedulres confluentes, con realce variable con el contraste, ADEMÁS de los hallazgos encefálicos específicos.
Comentarios
La definición propuesta requiere TANTO la ENCEFALOPATÍA* como el compromiso MULTIFOCAL (POLISINTOMÁTICO).
Un evento único de EMDA puede evolucionar durante un período de hasta 3 meses, con fluctuaciones clínicas, tanto en la intensidad de los síntomas y signos presentes, como en la aparición de nuevos.
El DIAGNÓSTICO de EMDA se debe fundamentar en la CLÍNICA ANTE TODO, los hallazgos de la RNM aislados NO SON SUFICIENTES para el diagnóstico.
Hallazgos de laboratorio típicos de la EMDA son:
aumento de las proteínas en LCR
GB aumentados en LCR, hasta mayores de 50 cel/mm3;
BOC ocasionales.
Diferencias entre EMDA/EM
La ENCEFALOPATÍA TÍPICAMENTE NO SE ASOCIA a EM.
El compromiso de la SG puede verse en la EMDA, pero es INFRECUENTE en la EM.
La PLEOCITOSIS es MUY ATÍPICA en la EM.
La presencia de BOC es mucho más frecuente en la EM que en la EMDA.
EMDA recurrente
Un nuevo episodio de EMDA que ocurre 3 o más meses después del inicio del primer evento, con recurrencia de los mismos signos y síntomas, SIN COMPROMISO DE NUEVAS ÁREAS tanto por la CLÍNICA, el EXAMEN o la RNM.
El nuevo evento NO debe ocurrir DURANTE el tratamiento con CORTICOIDES, así como tampoco debe ocurrir dentro del mes posterior a la supresión de los mismos.
La RM NO debe mostrar NUEVAS lesiones, aunque las lesiones originales pueden agrandarse.
No hay una mejor explicación etiológica del nuevo evento.
EMDA multifásica
1. Nuevo evento que cumple criterios de EMDA y que INVOLUCRA NUEVAS ÁREAS del SNC, evidenciado por la historia clínica, el examen neurológico y la RM.
Comentarios
La diferencia consiste en que si el segundo evento compromete nuevas áreas cerebrales “MULTIFÁSICA”, o si consiste en la “recaída” de las mismas áreas afectadas en el primer evento “RECURRENTE”.
Los nuevos eventos deben cumplir los criterios de EMDA.
La RM puede mostrar resolución completa o persistencia parcial de las lesiones.
Los casos con MÁS de dos episodios son ALTAMENTE SOSPECHOSOS de EM.
Los términos BIFÁSICA y RECAÍDA han sido abandonados para simplificar las definiciones.
Estudios con seguimiento prolongado (1-2 décadas), serán necesarios para comprobar la utilidad de estas definiciones.
Esclerosis múltiple pediátrica (infancia y adolescencia)
El diagnóstico de EM en la edad pediátrica requiere la presencia de episodios de desmielinización del SNC con separación en el tiempo y en el espacio al igual que en los pacientes adultos, sin una edad mínima estipulada.
Diseminación espacial
Tres o más lesiones periventriculares.
Una lesión yuxtacortical.
Una lesión infratentorial.
2. Dos lesiones en la RM, de las cuales una debe ser cerebral, sumado a BOC y/o elevación del IgG index en LCR, cumple criterio de diseminación espacial.
Un episodio que reúne los criterios para diagnóstico de EMDA no debe ser considerado un primer evento de una EM, a no ser que el curso evolutivo lo sugiera.
Al igual que en adultos, los niños con 2 episodios desmielinizantes aislados separados en tiempo y espacio, cumplen criterios de EM.
En la población pediátrica estos eventos NO DEBEN CUMPLIR CRITERIOS DIAGNÓSTICOS DE EMDA.
Un niño cuyo evento inicial configuró una EMDA, un segundo evento desmielinizante aislado (CIS) no se considera suficiente para el diagnóstico de EM.
El Consenso actual considera que se requiere evidencia adicional de diseminación espacial y temporal, ya sea clínica (tercer evento al menos 3 meses después del segundo), o en la RM (nuevas lesiones en T2 que aparecen al menos 3 meses luego del segundo evento).
Anexo 2 (4)
RM en etapa aguda. Subgrupos radiológicos
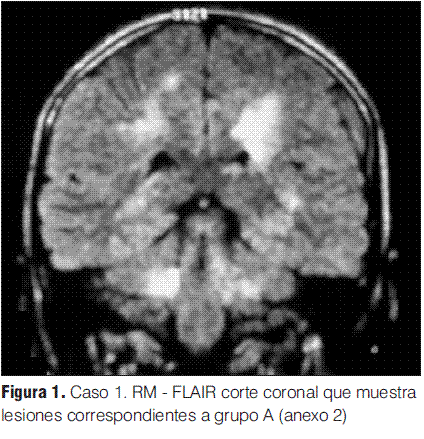
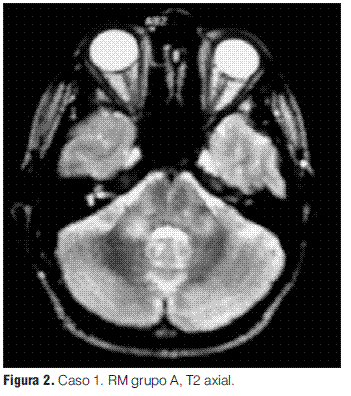
Grupo A: EMDA a pequeñas placas, lesiones menores a 5 mm de diámetro.
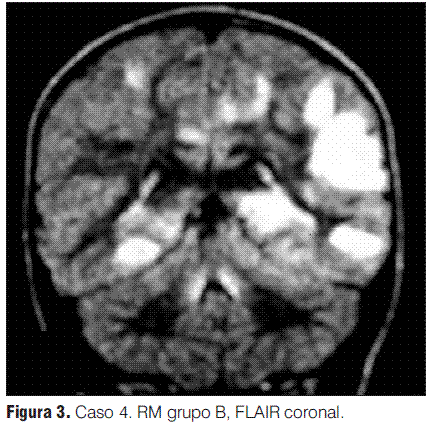
Grupo C: EMDA compromiso simétrico bitalámico además de las lesiones distribuidas en la sustancia blanca, que pueden ser pequeñas o grandes placas.
Grupo D: corresponde a la encefalomielitis hemorrágica aguda, con diferentes grados de hemorragia dentro o adyacente a las grandes placas desmielinizantes.
Puede existir compromiso simultáneo de la MÉDULA ESPINAL, con aumento de su calibre en T1 y señal hiperintensa en T2 y FLAIR, afectando frecuentemente varios segmentos medulares.
Escala expandida del estado de discapacidad de Kurtzke


{kind=link}