










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versão On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.77 no.3 Montevideo out. 2006
LOS EXPERTOS RESPONDEN
Arch Pediatr Urug 2006; 77(3): 308-312
Parálisis flácida en la infancia
Dr. Gabriel González Rabelino 1
1. Profesor Agregado de Neuropediatría.
¿Cuál es la definición o el concepto de parálisis flácida en el niño?
Es una definición operativa, enmarcada en la campaña de la Organización Mundial de la Salud (OMS), de la década de los ochenta, con el objetivo de erradicar la polio del mundo, por lo que intenta incluir todos los casos de probable poliomielitis.
Esta definición incluye tres grandes aspectos:
1. Instalación aguda o hiperaguda en menos de 5 días.
2. Cuadro clínico caracterizado por una disminución o pérdida de fuerzas y tono muscular (flacidez) de una o más extremidades (espinal), pudiendo acompañarse de participación craneal.
3. Presentarse en niños, menores de 15 años.
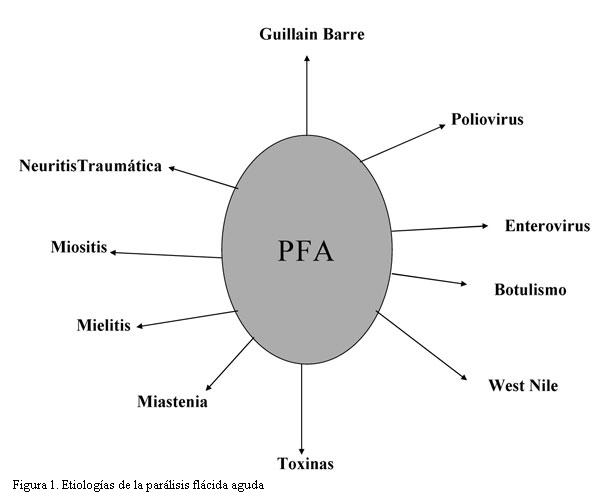
Esta definición abarca fundamentalmente las enfermedades del sistema nervioso periférico, que cursan con paresia e hipotonía y en las cuales se debe descartar obligatoriamente el virus de la polio, aunque como veremos existen múltiples etiologías (figura 1).
¿Cuál es la causa más común de parálisis flácida aguda (PFA) en la infancia?Todas las series muestran como principal causa al síndrome de Guillain Barré, en general con porcentajes que oscilan entre el 50 y 60% de las PFA. Las otras causas, menos frecuentes, incluyen mielitis transversa, polio vacunal, neuritis traumática, encefalomielitis, botulismo, miastenia y enfermedades por tóxicos (metales pesados, órganofosforados) o neurotoxinas como la provocada por picadura de garrapatas en otras regiones.
¿Cuál es la situación nacional y mundial
de la poliomielitis actualmente?
La incidencia en nuestro país de PFA, según el boletín de polio de la Organización Panamericana de la Salud (OPS), fue de 1,22/100.000 niños menores de 15 años en el año 2005 y de 1,59 en el presente año.
En nuestro país la última epidemia de polio virus salvaje fue en el año 1979, registrándose dos casos de polio vinculado a la vacuna en el año 1989 y 2002. En el continente americano el último caso de polio salvaje fue en 1991 en Perú, en la zona de Pichinaki.
A nivel mundial no se llegó a erradicar la polio del planeta, como se había planificado para el 2000, y la situación se redujo de más de 125 países con polio endémico en 1988 a menos de 6 en el año 2003 (Afganistán, Egipto, India, Nigeria, Pakistán, Níger); sin embargo, cuando se estaba próximo a la meta, en los últimos años más de 21 países libres de polio en estas regiones se volvieron a afectar, existiendo 92 casos en el año 2005 y hasta mayo del presente año 225 casos confirmados de polio salvaje.
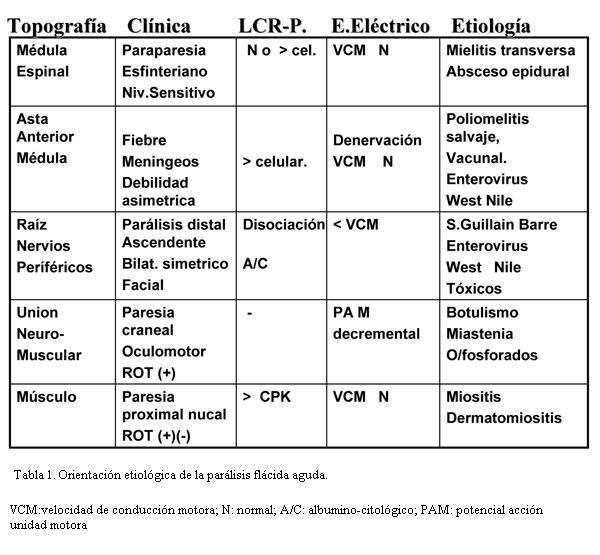
¿Cuáles son las características clínicas y paraclínicas más salientes de cada una de las causas de PFA?
La clínica, el líquido cefalorraquídeo y el estudio electrofisiológico nos permiten orientar el diagnóstico topográfico y etiológico ante una PFA y realizar el tratamiento oportuno y adecuado (tabla 2).
Luego de esta reseña sobre PFA, hablemos sobre su causa más frecuente, el síndrome de Guillain Barré (SGB). ¿Cómo se podría definir esta entidad?
Este término se aplica actualmente a un amplio espectro de polirradiculoneuropatías inflamatorias agudas, adquiridas, inmunológicamente mediadas, presumiblemente desencadenadas por una infección previa, con mecanismos neurofisiopatológicos variados (desmielinización, lesión axonal motora, o sensitivo-motora) con manifestaciones clínicas variadas (paresia, arreflexia, ataxia, síntomas sensitivos, autonómicos) con formas de presentación y severidad variable, y con evolución, en general monofásica y autolimitada.
¿Qué nos podría comentar sobre
el diagnóstico?
En primer lugar, el reconocimiento clínico puede ser sencillo en las formas típicas que se presentan en niños mayores o adolescentes y muy compleja en niños pequeños o con variantes atípicas.
Los pilares del diagnóstico son la clínica, el estudio del líquido cefalorraquídeo, estudios electrofisiológicos y actualmente se intentan incorporar estudios serológicos con anticuerpos.
Se presenta, en general, en niños sanos, con una infección digestiva o respiratoria, precediendo entre 1 y 3 semanas el inicio de la enfermedad.
Los criterios de Asbury incluyen:
- Síntomas y signos requeridos para el diagnóstico:
- Debilidad muscular progresiva de más de una extremidad.
- Ausencia de reflejos de estiramiento.
- Hallazgos clínicos que apoyan fuertemente el diagnóstico:
- La debilidad muscular se desarrolla rápidamente (gravedad máxima a las 4 semanas).
- Simetría relativa.
- Signos y síntomas sensitivos relativamente leves.
- Pares craneanos: VII par afectado en 50% (en menos del 5% puede iniciarse por pares craneanos).
- La recuperación comienza 2–4 semanas luego de haber alcanzado la gravedad máxima.
- Alteraciones autonómicas.
- Sin fiebre al comienzo de los síntomas neurológicos.
- Hallazgos en el LCR que apoyan fuertemente el diagnóstico:
- Proteínas elevadas después de la primera semana
- Células: no más de 10 leucocitos / mm3 (Hay autores que aceptan hasta 50 leucocitos / mm3).
- Hallazgos neurofisiológicos que apoyan fuertemente el diagnóstico:
- Velocidad de conducción (VC) con disminución > 60% en 80% de casos (no uniforme por característica segmentaria del proceso).
- Bloqueo de conducción en 80%.
- Aumento de latencias distales.
- Latencia de onda F aumentada o ausente.
Existen signos que deben hacer dudar del diagnóstico y otros que lo excluyen:
- Rasgos dudosos para el diagnóstico:
- Presencia de un nivel sensitivo.
- Marcada o persistente asimetría de los síntomas o de los signos.
- Disfunción esfinteriana persistente y grave.
- Más de 50 células/mm3 en el líquido cefalorraquídeo.
- Rasgos que excluyen el diagnóstico:
- Diagnóstico de botulismo, miastenia grave, poliomielitis o neuropatía tóxica.
- Trastornos en el metabolismo de las porfirinas.
- Difteria reciente.
- Síndrome sensitivo puro sin debilidad.
Es de destacar que los criterios diagnósticos de SGB no abarcan el espectro clínico completo de este trastorno.
Por lo tanto, el diagnóstico se debe basar en hallazgos clínicos, de laboratorio y electrofisiológicos consistentes y en la exclusión de otras condiciones con formas de presentación similar.
Aproximadamente el 10% de los casos no cumplen con estos criterios diagnósticos, son consideradas variantes atípicas de Guillain Barré (Ropper), las cuales pueden ser:
Formas regionales:
- Síndrome de Fisher (ataxia, oftalmoplejia y arreflexia).
- Cérvico-faringo-braquial (frecuentemente ptosis).
- Parálisis oculofaríngea.
- Predominio de la paraparesia.
- Parálisis facial bilateral con parestesias distales.
- Oftalmoplejia con Ac. GQ1b.
Formas funcionales:
- Ataxia sin disartria o nistagmo.
- Forma sensitiva pura.
- Pandisautonómica.
- Forma motora pura.
- Forma axonal.
Los rasgos clínicos variantes pueden ser fiebre al inicio, pérdida sensitiva grave, progresión luego de las cuatro semanas, cese de la progresión sin recuperación, alteración de los esfínteres (puede haber una paresia vesical transitoria) o afectación del SNC; a nivel del líquido cefalorraquídeo puede faltar el aumento de las proteínas o presentar entre 11 a 50 mononucleares por mm3. Si bien la presencia de uno de estos hallazgos no descarta el diagnóstico de SGB, debe plantear dudas y la presencia de dos variantes hacer sospechar otro diagnóstico.
¿Cuáles son las dificultades habituales del diagnóstico en el niño a las que se enfrenta el pediatra?
Los síntomas iniciales pueden ser difíciles de interpretar, entre el 50 y 80% de los niños tendrán dolor significativo al inicio, siendo difícil distinguir en el niño pequeño, entre paresia e inmovilidad por dolor, por lo que es habitual que en estos casos se sospechen enfermedades músculo–esqueléticas, como miositis, traumatismos o artralgias.
En segundo lugar, casi la mitad de los pacientes se presentan como “atáxicos”, por una falsa ataxia por paresia o una verdadera ataxia cerebelosa o sensitiva, en estos casos se puede confundir con intoxicaciones o cerebelitis.
En tercer lugar la irritabilidad, somnolencia y signos meníngeos pueden hacer sospechar una meningitis, encefalitis o tumor cerebral.
Quisiera destacar, además de las variantes controvertidas del síndrome con participación del SNC, que la presencia de un compromiso esfinteriano, reflejos vivos o signo de Babinsky deben obligar a descartar una mielopatía (compresión medular, mielitis) y realizar una RNM del raquis en forma urgente.
¿Cuál es el valor de realizar la punción lumbar?
Uno de los grandes aportes de Georges Charles Guillain y Jean Alexander Barré, fue describir en el líquido cefalorraquídeo el aumento de proteínas sin elevación de la celularidad, que fue crucial para diferenciar esta condición de entidades que hacían estragos en la época (sífilis, tuberculosis, poliomielitis).
El clásico patrón del estudio citoquímico del líquido cefalorraquídeo con aumento de las proteínas y recuento celular bajo, se denomina disociación albúmina citológica, con proteínas mayores de 45 mg/dl y menos de 10 células, que característicamente se observa después de la primera semana. Por lo tanto, el estudio de líquido cefalorraquídeo normal en la primera semana no descarta el diagnóstico y es útil para alejar otros diagnósticos diferenciales que cursan con pleocitosis (polio, otros enterovirus, mielitis, VIH, Lyme, entre otros).
Por lo tanto en estos casos debe ser reiterado luego de los 10 días.
Aproximadamente un 10% de los casos pueden no tener hiperproteinorraquia, lo cual se explicaría porque la inflamación o desmielinización es distal a las raíces nerviosas.
¿Qué valor tienen los estudios electrofisiológicos?
Como mencionamos previamente, son uno de los pilares del diagnóstico y nos permiten tipificar en forma precisa si la lesión de los nervios es de tipo desmielinizante (con enlentecimiento marcado de la velocidad de conducción o con bloqueo) o axonal (con amplitudes motoras y eventualmente sensitivas disminuidas con valores normales o ligeramente enlentecidos de latencias y velocidad de conducción), lo cual tendrá implicancias pronósticas.
Esto nos permite clasificar el SGB en: 1) Polirradiculoneuropatía inflamatoria desmielinizante aguda (AIDP) que es la forma clásica que predomina en occidente, donde el blanco inmunológico está entre la célula de Schwann y la mielina, con un pronóstico en general excelente. Otras variantes regionales como Miller Fisher también presentan este patrón eléctrico. 2) Neuropatía axonal motora aguda (AMAN) y neuropatía axonal sensitivo–motora aguda (AMSAN) con un blanco inmunológico en los nódulos de Ranvier.
La neuropatía axonal motora aguda es más frecuente en niños que adultos y se presenta vinculada a la infección por Campylobacter jejuni, especialmente en China, Japón y algunos países de América latina. En estas formas pueden mantenerse los reflejos y las disfunciones autonómicas son raras. El pronóstico de esta forma es variable, con formas de recuperación pobre y lenta y otros con recuperación rápida.
La neuropatía axonal sensitivo-motora aguda (AMSAN) se observa fundamentalmente en adultos y son formas más graves y de peor pronóstico.
En nuestro medio tenemos una gran limitación en dichos estudios, recientemente el Dr. Cerisola realizó una revisión de los últimos 6 años y solamente en el 33% de los casos se realizó el estudio electrofisiológico, con informes insuficientes.
En cuanto al tratamiento ¿qué aspectos resaltaría?
En primer lugar la hospitalización con monitoreo de la función respiratoria, hasta que por lo menos se haya establecido el máximo grado de discapacidad.
La analgesia muchas veces no es jerarquizada, debiendo recurrir inicialmente a analgésicos y fisioterapia pasiva; de no tener respuesta se podrán usar antineuríticos (gabapentina, carbamazepina) y eventualmente opiáceos con cuidadosa monitorización de los efectos adversos.
La monitorización continua de la frecuencia cardíaca y presión arterial es fundamental para prevenir la muerte por arritmias e inestabilidad hemodinámica. La asistencia ventilatoria mecánica está indicada cuando la capacidad vital desciende por debajo de 15 ml/kg o ante la instalación de una parálisis bulbar.
La inmunoterapia está indicada cuando el paciente no puede caminar independientemente (grado 3 de escala Hughes), o cuando presenta compromiso bulbar con riesgo de aspiración, no siendo efectivo luego de las 3 a 4 semanas de iniciados los síntomas.
Los trabajos extrapolados en adultos y las series pequeñas en niños con ensayos clínicos europeos y norteamericanos han demostrado la eficacia de la plasmaféresis y las inmunoglobulinas intravenosas en acelerar la recuperación y evitar progresión de la enfermedad. La Academia Americana de Neurología recomienda el uso de plasmaféresis o inmunoglobulinas en niños con formas severas de SGB, no recomendando la asociación de los dos tratamientos ni el uso de corticoides. Actualmente la mayoría de los centros prefieren el uso de inmunoglobulinas en niños, por su mayor facilidad de aplicación y seguridad.
Los planes terapéuticos se basan en la administración de 2 g/kg en cinco días o en dos días; si bien este último plan podría ser más efectivo, puede producir mayores efectos adversos.
¿Cuál es el pronóstico del SGB en niños?
Aproximadamente el 90% de los niños se recuperan completamente entre los 6 y 12 meses de iniciada la enfermedad, y los que no se recuperan completamente caminan en forma independiente y tienen sólo secuelas menores.
Las recaídas son raras en niños y se ven en menos del 10%, pudiendo estos casos beneficiarse de un nuevo tratamiento breve.
Son factores de mal pronóstico: la discapacidad máxima al inicio de la enfermedad, necesidad de AVM, compromiso craneal, y formas axonales.
La mortalidad pediátrica oscila alrededor del 5% y es consecuencia de insuficiencia respiratoria, disautonomía y sepsis, fundamentalmente.
Referencias bibliográficas
Buompadre MC, Gañez LA, Miranda M, Arroyo H. Variantes inusuales del Síndrome de Guillain-Barré en la infancia. Rev Neurol 2006; 42: 85-90.
Cerizola A. Síndrome de Guillain-Barré en pediatría [monografía postgrado]. Montevideo: Facultad de Medicina, jul 2006.
Hughes RAC, Cornblath DR. Guillain Barré syndrome. Lancet 2005; 366: 1653-66.
Korinthenberg R, Schessi J, Kirschner J. Intravenously administered immunoglobulin in the treatment of childhood Guillain-Barré syndrome: a randomized trial. Pediatrics 2005; 116: 8-14.
Papazian O, Alfonzo I. Polirradiculoneuropatías autoinmunes agudas. Rev Neurol 2002; 34: 169-77.
Pascual-Pascual SI. Aspectos actuales de las neuropatías inflamatorias agudas y crónicas. Síndrome de Guillain Barré y polineuritis crónica inflamatoria desmielinizante. Rev Neurol 2002; 35: 269-76.
Sladky JT. Guillain-Barré Syndrome in Children. J Child Neurol 2004; 19: 191-200.
Tellería-Díaz A, Calzada-Sierra DJ. Síndrome de Guillain Barré. Rev Neurol 2002; 34: 966-76.
Correspondencia: Dr. Gabriel González Rabelino
República Argentina, manzana 7, solar 19 (Shangrilá)
Ciudad de la Costa, Uruguay.
E-mail: viciogon@hotmail.com


{kind=link}
{kind=link}