










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versão On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.76 no.2 Montevideo jun. 2005
CASO CLÍNICO
Arch Pediatr Urug 2005; 76(2): 135-139
Linfangiomatosis pulmonar difusa.
Una causa poco frecuente de insuficiencia respiratoria en el niño
DRAS. ALICIA FERNÁNDEZ 1, ÁNGELES RODRÍGUEZ 2, CARMEN GUTIÉRREZ 3, MARTA ALBERTI 4
1. Pediatra intensivista. Ex -Profesora Adjunta de Cuidados Intensivos de Niños. UCIN. Centro Hospitalario Pereira Rossell
2. Patóloga Pediatra. Laboratorio de Patología Pediátrica. Centro Hospitalario Pereira Rossell.
3. Jefe del Laboratorio de Patología Pediátrica. Centro Hospitalario Pereira Rossell.
4. Profesora Titular de Cuidados Intensivos de Niños.
UCIN. Centro Hospitalario Pereira Rossell
Fecha recibido: 19 de mayo de 2005
Fecha aprobado: 13 de julio de 2005
Resumen
Se presenta un caso de linfangiomatosis pulmonar difusa en una paciente de 12 años con historia de varios meses de evolución caracterizada por disnea y adelgazamiento y en la que se constatan infiltrados pulmonares y derrame pleural bilateral. El estudio histológico postmortem mostró una lesión vascular pulmonar de naturaleza linfática, consistente en una proliferación de estructuras vasculares de fina pared y contenido luminal sin sangre, localizada en septos interlobulares, bronquios, adventicias vasculares y pleuras. La linfangiomatosis pulmonar es una lesión rara de pronóstico pobre.
Palabras clave: PULMÓN-patología
Summary
A 12 years old girl presented with dyspnea, weight loss, pulmonary infiltrates and pleural effusions. The lung lesion as seen at the necropsy consisted of a lymphatic vascular proliferation limited to interstitial septae, bronchi, adventitia of large vessels and pleura. Disseminated lymphangiomatosis is a rare disorder with a poor prognosis.
Key words: LUNG-pathology
Introducción
Las patologías linfáticas primarias del pulmón son raras y difíciles de identificar. Se reconocen por lo menos cinco entidades clinicopatológicas bien definidas: el linfangioma, las linfangiectasias, la linfangioleiomiomatosis, la linfangiomatosis pulmonar difusa y el compromiso pulmonar en el marco de una linfangiomatosis sistémica. En opinión de expertos (1), el análisis de la literatura muestra una gran confusión terminológica. La clasificación de Hilliard (2) ha contribuido a delimitar el tema con mayor claridad. El sistema linfático pulmonar puede además ser secundariamente afectado en el curso de otras enfermedades como neoplasias, traumatismos y cirugía.
La linfangiomatosis pulmonar difusa es una enfermedad rara, de causa desconocida, que afecta básicamente a niños y adolescentes, comprometiendo por igual a ambos sexos. Se presenta clínicamente con tos seca, disnea progresiva, broncoespasmo y derrames en general quilosos. Su prevalencia no está claramente determinada, posiblemente por el desconocimiento general sobre esta entidad, lo que retarda el diagnóstico.
No hay hasta el momento un tratamiento curativo. Se han ensayado diferentes protocolos; algunos han tenido buen resultado en lo inmediato pero sin mejoría a largo plazo. El transplante pulmonar es una alternativa válida.
Caso clínico
Niña de 12 años, raza blanca. Sin antecedentes personales patológicos. Inmunizada. Buen crecimiento y desarrollo. Comienza varios meses previos al ingreso con repercusión general, astenia, anorexia, adinamia y adelagazamiento. En las horas previas tos seca persistente, quejido, dolor tipo puntada en hemitórax derecho, polipnea y disnea. Radiografía de tórax: opacidad homogénea en hemitórax derecho con ocupación pleural líquida. Toracocentesis diagnóstica: 1.500 ml de líquido serohemático con las características de un trasudado. Cultivos negativos. No se deja drenaje. Se indican antibióticos intravenosos. Mala evolución, peoría clínica. Radiografía: compromiso bilateral y neumotórax izquierdo. Toracocentesis bilateral: 300 ml de líquido serohemático a derecha y 600 ml de líquido y aire a izquierda. Drenaje bilateral. Se traslada a la UCI en el postoperatorio inmediato. Al ingreso: lúcida, ventilación espontánea con máscara de flujo libre, saturometría de pulso 95%. Polipnea 48 pm, tiraje generalizada con retracción, balanceo. Síndrome de condensación a bronquio permeable en 2/3 inferiores de ambos hemitórax. Gasto de 300 ml por cada drenaje con las características descritas. Taquicardia regular 130 pm, PA 130/60/50 mmHg, pulsos presentes en los cuatro miembros y tiempo de recoloración inmediato. No visceromegalias.
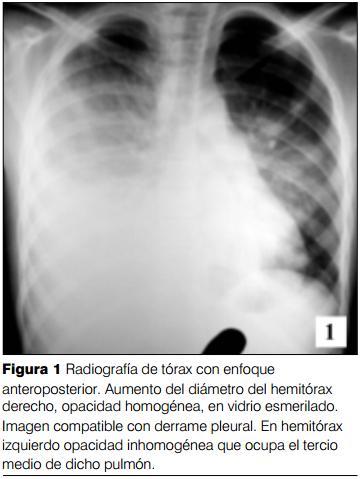
Impresión clínica: desnutrición severa. Postoperatorio inmediato de toracocentesis y drenaje bilateral. Neumonía extensa comunitaria a patógeno no identificado con derrame serohemático. Mala evolución pese al tratamiento instituido.
Conducta: 02, hidratación endovenosa. Se mantienen vancomicina y ceftriaxona intravenosas y se agrega claritromicina vía oral. Hemograma: glóbulos blancos 11.300 elementos/mm3, 78% neutrófilos, hemoglobina 7 g/l, hematocrito 23%, PCR 80, VES 99 mm. Gases en sangre: acidosis respiratoria sin hipoxemia.
Permanece en UCI 28 días. Mantiene síndrome funcional respiratorio, sin hipoxemia. El gasto por los drenajes disminuye. Se retiran. Mantiene compromiso difuso parenquimatoso y pleural bilateral, sin anomalías específicas en la radiografía y tomografía.
Hemocultivos, cultivos de líquido pleural, baciloscopías, PCR para bacilo de Koch, PPD, investigación micológica, parasitaria, neoplásica y HIV negativos.
Dosificación de inmunoglobulinals y complemento normales. Anticuerpos antinucleares, antimitocondriales, antimúsculo liso y células LE negativos. Estudio con lámpara de hendidura normal. Biopsia pleural y pulmonar no concluyente por el material insuficiente de la muestra.
Con planteo de mesenquimopatía se inicia metilprednisolona, que se mantiene 15 días. Requiere nutrición parenteral y enteral por sonda nasogástrica. Febrícula persistente, cultivos negativos. Reactantes de fase aguda: elevados. A los 21 días del plan inicial de antibióticos se rota a meropenem. Pasa a sala general para continuar estudio y tratamiento, alimentándose por vía enteral, en apirexia con polipnea leve, ventilando espontáneamente con cánula nasal con 3 litros de 02 y 26 kg de peso.
A los seis días reingresa a la UCI por exacerbación del síndrome funcional respiratorio, tos persistente que le impide hablar e insuficiencia respiratoria. Se inicia apoyo ventilatorio mecánico invasivo. El deterioro progresivo de la función pulmonar y la reinstalación del derrame pleural determinan toracotomía. En la misma se encuentran tejidos necróticos. Sangrado masivo. No se logra hemostasis. Fallece. Diagnóstico clínico: adolescente de 12 años; neumonía extensa bilateral comunitaria a patógeno no identificado con insuficiencia respiratoria grave y derrame bilateral hemático no purulento de etiología no determinada. Sangrado pulmonar masivo.
Anatomía patológica
Se realizó autopsia parcial limitada a tórax (de acuerdo a la autorización obtenida). Se constataron sinequias pleurales bilaterales extensas y líquido con tinte hemorrágico. Las pleuras estaban gruesas y el parénquima pulmonar aumentado de consistencia. Peso pulmonar derecho 460 g; pulmón izquierdo 250 g. El estudio histológico mostró una proliferación vascular linfática de caracteres típicos localizada en las rutas habituales de los linfáticos pulmonares en forma difusa en ambos pulmones (figura 2). Se constató un compromiso de septos interlobares, interlobulillares, pleuras y tejido conectivo peribronquial. Las luces de las estructuras vasculares estaban vacías o contenían material proteináceo. Se observó escasa celularidad entre las estructuras vasculares, constituidas por células fusiformes las que con inmunoperoxidasa fueron actina +, desmina + y vimentina +.
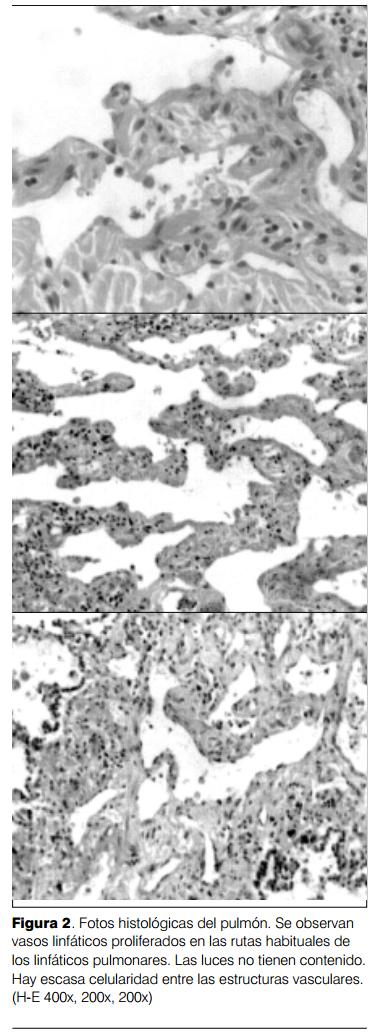
Diagnóstico anátomo patológico: linfangiomatosis pulmonar difusa.
Discusión
Las enfermedades linfáticas que afectan difusamente el parénquima pulmonar determinan dificultades al clínico. La incidencia de esta enfermedad es baja, planteándose otras enfermedades habituales y realizándose tratamientos que no son los indicados. La no respuesta terapéutica específica para determinados planteos diagnósticos y la prolongación del cuadro, obliga a la realización de la biopsia pulmonar, siendo fundamental la obtención de material adecuado para el análisis histopatológico, que será en definitiva el que permita llegar al diagnóstico.
En el caso presentado la imagenología no fue concluyente y la biopsia fue insuficiente; no se llegó al diagnóstico de la enfermedad en vida de la paciente. El diagnóstico se realizó por la autopsia.
La linfangiomatosis pulmonar difusa (1) se caracteriza por afectar la totalidad del pulmón, presentar un aumento en el número de los canales linfáticos que están localizados en las rutas habituales de los linfáticos pulmonares y concomitantemente una proliferación leve de células fusiformes de bordes romos. Estos canales vasculares tienen luces ocupadas por material proteináceo, sin sangre. Las células fusiformes se disponen en paralelo entre los vasos y tienen carácter maduro. Con inmunohistoquímica se identifica positividad para vimentina, actina y desmina, lo que las vincula con las células musculares lisas. Con CD31 se demuestra el carácter linfático del endotelio vascular proliferado (3). Se pueden observar colecciones de macrófagos cargados de hemosiderina en el parénquima pulmonar adyacente.
La lesión se observa en el niño, el adolescente y el adulto joven. Se ha estimado que pueda constituir hasta 4% de los casos de enfermedad pulmonar intersticial crónica en el niño (4).
Desde el punto de vista imagenológico (5,6) se observa en la radiología y en la tomografía convencional una infiltración de las partes blandas mediastinales, engrosamiento de los septos interlobares, derrame pleural y engrosamiento pleural que en ocasiones puede estar calcificado. Hay engrosamiento peribroncovascular e intersticial. Algunos pacientes tienen opacidades en vidrio esmerilado e infiltración perihiliar. En los casos de linfangiomatosis sistémica con compromiso pulmonar hay quistes que acompañan opacidades lineares en los pulmones (6).
El diagnóstico diferencial desde el punto de vista anátomo-patológico debe plantearse con: linfangiectasia, linfangiomiomatosis, hemangiomatosis, linfangioma, sarcoma de Kaposi y el hemangioendotelioma kaposiforme.
El término linfangiectasia debería reservarse para los casos de lesiones congénitas o adquiridas en las que hay dilatación de canales linfáticos sin aumento numérico (1-3).
La linfangioleiomiomatosis es una lesión que compromete a mujeres en edad reproductiva. El proceso lesional se extiende más allá de las rutas linfáticas comprometiendo el parénquima alveolar (1,3,6-10). Hay una densa proliferación de células musculares lisas de aspecto irregular e inmaduro, que pueden rodear e infiltrar paredes vasculares. Los alvéolos tienen aspecto quístico rodeado por bandas de células musculares lisas. La entidad tiene una forma esporádica y otra vinculada genéticamente a la esclerosis tuberosa (10). Es importante no confundir a la linfangiomatosis pulmonar difusa con esta entidad ni con las linfangiectasias (11).
La hemangiomatosis pulmonar es otro diagnóstico diferencial a considerar (1,6-8). Muchos de los casos publicados tienen similitud histológica con la linfangiomatosis pulmonar, pero con contenido sanguíneo en las luces vasculares. Se reconoce una forma de hemangiomatosis pulmonar de tipo capilar (12-14) que se asocia a hipertensión arterial (en adultos). La proliferación capilar se localiza en paredes alveolares y de paredes de vasos pulmonares, vía aérea y pleura. No muestra predilección por rutas linfáticas y no se asocia a proliferación de músculo liso.
Los linfangiomas en general son masas tumorales que en muchos casos pueden resecarse.
En muchos de los casos descritos de linfangiomatosis pulmonar, el mediastino está además comprometido (6,16,17). Por esa razón se ha considerado que esta entidad debe estar considerada en el diagnóstico diferencial de las masas mediastinales con lesiones pulmonares. Es importante excluir a los efectos terminológicos las formas diseminadas o sistémicas con compromiso múltiple extrapulmonar (17).
Las terapéuticas propuestas hasta ahora ofrecen una pobre respuesta y la evolución a una insuficiencia respiratoria progresiva y a la muerte es lo que en general acontece a corto o mediano plazo. Se han utilizado en el tratamiento corticoides y citostáticos (18), interferón alfa (16,17,19,20) y radioterapia.
Conlusión
La necropsia constituye una maniobra semiológica en el paciente fallecido, siendo su realización un modo de evaluar la calidad asistencial. En esta historia de una adolescente de 12 años con una enfermedad pulmonar que fue catalogada como una neumonía atípica con insuficiencia respiratoria en la evolución, la clínica no permitió llegar al diagnóstico de la enfermedad de base en vida. De acuerdo con los criterios de Goldman (21) al no figurar el diagnóstico definitivo al egreso y ser un hallazgo de la necropsia, se clasifica como error tipo II. Es decir, diagnóstico que de haberse realizado en vida no hubiese supuesto un cambio en la sobrevida de acuerdo a la terapéutica con evidencia demostrada en esta enfermedad. El diagnóstico obtenido por la autopsia es fundamental para el aprendizaje del grupo asistencial y la devolución de la información a los padres.
Citas bibliográficas
1. Tazelaar H, Kerr D, Kousem S, Saldana HJ, Langston C. Diffuse pulmonary lymphangiomatosis. Hum Pathol 1993; 24: 1313-22.
2. Hilliard R, Kendry JB. Congenital abnormalities of the lymphatic system: a new clinical classification. Pediatrics 2000; 86: 988-94.
3. Jones KD, Colby TV. Developmental and pediatric lung disease. En: Leslie KO, Wick MR, eds. Practical pulmonary pathology. Philadelphia: Churchill Livingstone, 2005: 61-2.
4. Fan LL, Mullen AL, Brugman SM, Inscore SC, Parks DP, White CW. Clinical spectrum of chronic interstitial lung disease in children. J Pediatr 1992; 121 (6): 867-72.
5. Swensen SJ, Hartman TE, Mayo JR, Colby TV, Tazelaar HD, Muller NL. Diffuse pulmonary lymphangiomatosis: CT findings. Comput Assist Tomogr 1995; 19 (3): 348-52.
6. Yekeler E, Durusun M, Yildirim A, Tunaci M. Diffuse pulmonary lymphangiomatosis: imaging findings. Diag Interv Radiol 2005; 11: 31-4.
7. Pinkard NB. Lymphangioleiomyomatosis. En: Cage PT, ed. Diagnostic Pulmonary Pathology. New York: Marcel Decaer AG, 2000: 352-5.
8. Faul JL, Berry GJ, Colby TV, Ruoss SJ, Walter MB, Rosen GD, et al. Thoracic lymphangiomas, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndrome. Am J Respir Crit Care Med 2000; 161 (3 Pt 1): 1037-46.
9. Bittmann I, Rolf B, Amann G, et al. Recurrence of lymphangioleiomyomatosis after single lung transplantation: new insight into pathogenesis. Hum Pathol 2003; 34: 95-8.
10. Fukuoka J, Leslie KO. Chronic diffuse lung diseases. En: Leslie KO, Wick MR, eds. Practical pulmonary pathology. Philadelphia: Churchill Livingstone, 2005: 239-43.
11. Katzsenstein AL, Askin F. Diffuse pulmonary lymphangiomatosis. En: Katzenstein AL, ed. Katzenstein and Askin's Surgical Pathology of Non-Neoplastic Lung Disease. Philadelphia: Saunders, 1997: 386-7.
12. Tron V, Magee F, Wright JL. Pulmonary capillary hemangiomatosis. Hum Pathol 1986; 17: 1144-50.
13. Varnholt H, Kradin R. Pulmonary capillary hemangiomatosis arising in hereditary hemorrhagic telangiectasia. Hum Pathol 2004; 35: 266-8.
14. Churg A, Wright JL. Pulmonary capillary hemangiomatosis. En: Leslie KO, Wick MR, eds. Practical pulmonary patology. Philadelphia: Churchill Livingstone, 2005: 396-9.
15. Takahashi K, Takahashi H, Maeda K, Homma S, Uekusa T, Dambara T, et al. An adult case of lymphangiomatosis of the mediastinum, pulmonary interstitium and retroperitoneum complicated by chronic disseminated intravascular coagulation. Eur Respir J 1995; 8(19): 1799-802.
16. Alvarez OA, Kjellin I, Zuppan CW. Thoracic lymphangiomatosis in a child. J Pediatr Hematol Oncol 2004; 26: 136-41.
17. Laverdiere C, Dubois DM, Russo P. Improvement of disseminated lymphangiomatosis with recombinant interferon therapy. Pediatr Pulmonol 2000; 29(4): 321-4.
18. Canny GJ, Cutz E, MacLusky IB, Levison H. Diffuse pulmonary angiomatosis. Thorax 1991; 46(11): 851-3.
19. Reinhart MA, Nelson SC, Sencer SF, Bostrom BC, Kurachek SC, Nesbit ME. Treatment of childhood lymphangiomas with interferon-alpha. J Pediatr Hematol Oncol 1997; 19: 232-6
20. Rostem A. Treatment of thoracic lymphangiomatosis. Arch Dis Child 2000; 83: 138-9.
21. Goldman L, Sayson R, Robbins S, Cohn LH, Bettmann M, Weisberg M. The value of the autopsy in three medical eras. N Engl J Med 1983; 308 (17):1000-5.
Correspondencia: Dra. Alicia Fernández
Asamblea 4126. CP 11400
Email: alimarfer@hotmail.com

