










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versão On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.74 no.2 Montevideo ago. 2003
Enfermedad de Kawasaki
DR. JAVIER PREGO PETIT 1
1. Ex Profesor Adjunto de Pediatría. Coordinador del Departamento de Emergencia Pediátrica
Centro Hospitalario Pereira Rossell. Departamento de Emergencia Pediátrica.
Recibido: 7/5/03
Aceptado: 15/7/03
Referencia histórica
La primera descripción de la enfermedad de Kawasaki (EK) fue realizada en Japón por el Dr. Tomisaku Kawasaki en el año 1961. Se trataba de un niño de cuatro años, que ingresó al Hospital de la Cruz Roja en el sexto día de evolución de la enfermedad, que presentaba fiebre, adenopatía cervical, erupción cutánea, congestión conjuntival, alteraciones en labios y boca, ictericia, anemia hemolítica y descamación. Fue tratado con corticoides y antibióticos, siendo dado de alta a los 41 días de iniciada la enfermedad. Fue considerada como una enfermedad de etiología desconocida, benigna, autolimitada, sin secuelas (1). En el año 1962, T. Kawasaki realizó una comunicación sobre siete casos clínicos, denominándolos como "síndrome febril no-escarlatina con descamación" y en el año 1964 comunicó 22 casos clínicos más, denominándolos como "síndrome mucocutáneo ocular" (1). A pesar de la acumulación de nuevos casos, muchos clínicos consideraban que no se trataba de una nueva enfermedad, sino de una presentación atípica del síndrome de Stevens-Johnson. En el año 1965, el Dr. Noburu Tanaka realizó una autopsia a un niño que tenía el diagnóstico de síndrome mucocutáneo ocular, que había muerto en forma súbita e inesperada, descubriendo una trombosis en una arteria coronaria (1). Hasta ese momento no se había correlacionado la enfermedad con afectación de las arterias coronarias, y muchos clínicos negaron la asociación del síndrome mucocutáneo ocular con complicaciones cardíacas fatales. La primera publicación de la EK fue en el año 1967 (2), en idioma japonés, por T. Kawasaki, publicada en una revista de alergia, refiriendo 50 casos clínicos y estableciendo los criterios diagnósticos de la enfermedad. Esta publicación generó controversias en el ambiente médico japonés, sobre todo en lo relacionado al vínculo de la enfermedad con la afectación cardíaca. El Dr. Takajiro Yamamoto, en el año 1968, publicó una serie de casos de EK, en donde sobre 23 pacientes, once (48%) presentaban anormalidades electrocardiográficas, estableciéndose que la afectación cardíaca era un hecho común en esta enfermedad (3). La primer publicación en idioma inglés de EK fue realizada por T. Kawasaki en el año 1974, en la revista Pediatrics (4), denominándose como "síndrome mucocutáneo ganglionar". En el año 1970 se estableció por parte de Tanaka y colaboradores, al igual que otros autores japoneses (1), el vínculo entre las formas fatales de la poliarteritis nodosa infantil y las formas severas de la EK, considerándolas diferentes manifestaciones de una misma enfermedad, lo que fue reafirmado en el año 1977 por Landing y Larson (5), manifestando que estas dos enfermedades son indistinguibles desde el punto de vista anátomo-patológico.
Las primeras observaciones de la EK en Estados Unidos de Norteamérica (EE.UU.) fueron realizadas en la década de los 70 en Hawai, por parte de dos jóvenes médicos; Marian Melish, especialista en enfermedades infecciosas y Raquel Hicks, pediatra reumatóloga; aunque no establecieron el vínculo con la EK hasta el año 1973, al ver fotos de pacientes con EK procedentes de Japón, habiéndolos considerado previamente como casos de fiebre de origen desconocido con resolución espontánea (1). En el año 1976 publicaron una serie de pacientes hawaianos con EK, siendo la primera publicación de pacientes estadounidenses, la mayoría de origen asiático (6).
La razón del reconocimiento simultáneo en Japón y EE.UU. de esta enfermedad entre los años 1960 y 1970 continúa siendo desconocida, planteándose diferentes posibilidades
Una de ellas es que la EK sea una nueva enfermedad que emergió en Japón diseminándose a occidente a través de Hawai, en donde la enfermedad prevalecía en niños asiáticos. Otra alternativa es que la EK y la poliarteritis nodosa infantil formen parte de un espectro de una misma enfermedad, y que las formas clínicas moderadas de la EK estuvieran enmascaradas entre otras enfermedades, como la escarlatina durante la era preantibiótica. Es probable que la mejoría en el nivel de la asistencia médica, particularmente la utilización de antibióticos para el tratamiento de infecciones bacterianas productoras de toxinas, redujera el número de enfermedades febriles que cursan con erupción cutánea, lo que llevó a reconocer a la EK como una entidad clínica diferente (1). Recientemente se encontró en Londres, en el hospital St. Bartholomew´s, el corazón conservado de un niño de siete años que murió en el año 1870 por "hidropesía por escarlatina". El corazón presentaba tres aneurismas en las arterias coronarias con trombosis, hallazgos compatibles con EK (7).
En el año 1978 se publicaron en EE.UU., por parte del Centro de Control y Prevención de Enfermedades, los criterios diagnósticos de la EK, modificando los originales de T. Kawasaki (8). Después de la publicación de la eficacia de la inmunoglobulina de uso intravenoso (IGIV) para el tratamiento del púrpura trombocitopénico idiopático, se comenzó a utilizar en Japón este régimen terapéutico para la EK (1). En el año 1988 la Academia Americana de Pediatría (AAP) aprobó el uso de IGIV más ácido acetilsalicílico (AAS) (9) y en el año 1993 la American Heart Association (AHA) publicó los criterios diagnósticos y guía terapéutica de la EK (10). Más recientemente, se propusieron nuevos tratamientos con esteroides y agentes antiinflamatorios para aquellos casos de EK que no responden al tratamiento clásico (11,12).
El primer caso clínico nacional fue publicado en el año 1991 (13).
Epidemiología
La EK afecta primariamente a niños pequeños y tiene una considerable variabilidad geográfica y racial. La EK es más frecuente en menores de 4 a 5 años, siendo poco frecuente por debajo de los tres meses (14), aunque puede presentarse en todas las edades, desde la etapa neonatal (15) a la adolescencia (16). Los varones son más afectados que las niñas, con una relación de 1,4 a 1 (14). La incidencia de la EK es variable según diferentes países. En Japón es de 108 casos por 100.000 niños menores de cinco años de edad (17), siendo el país de mayor incidencia. En EE.UU. es de 10 casos por 100.000 niños menores de cinco años de origen no asiático y de 44 casos por 100.000 en los de origen asiático (7). En China la incidencia es similar a la de EE.UU. (18). En Australia la incidencia es de 3,7/100.000 en menores de cinco años, en las Islas Británicas es de 3,6/100.000 menores de cinco años (19) y en Chile 3/100.000 en menores de cinco años (20).
La mortalidad por EK también es variable según diferentes países. Japón presenta los valores más bajos, descendiendo de 1% en 1974 a menos de 0,1% en 1995 (17), en tanto en otros países es más elevada. En Inglaterra la mortalidad es de 3,7% (20). Esta diferencia en la mortalidad está vinculada a un mejor reconocimiento de la EK y por ende a un tratamiento precoz y adecuado, en los países en donde EK es más frecuente. La mortalidad es mayor en los varones y en los menores de un año de vida (21).
En EE.UU., la EK es más frecuente en las clases socioeconómicas medias y altas (22).
A pesar de un mayor conocimiento de EK, la epidemiología continúa evolucionando, pues aún se denuncia en diferentes partes del mundo el retardo en el diagnóstico y tratamiento. El Centers for Disease Control de EE.UU. mantiene sólo una vigilancia pasiva de la enfermedad y en general la medicina occidental no ha tenido la misma reacción que frente a otros problemas emergentes, probablemente a causa de la diversidad y complejidad de los datos, muchas veces controversiales que surgen de la experiencia con EK (20).
En los países en desarrollo, la EK ha sustituido a la fiebre reumática aguda como la causa más frecuente de cardiopatía adquirida en los niños (23).
Etiología
La etiología de la EK continúa siendo desconocida. Sin embargo, diversos aspectos clínicos y epidemiológicos sugieren una etiología infecciosa. Al ser una enfermedad autolimitada, acompañada de fiebre, exantema, enantema y adenopatía, sugiere una causa infecciosa. Es una enfermedad que tiene un patrón de incidencia estacional, con mayor predominio durante los meses de primavera e inicio del verano, como sucede en las infecciones virales. El desarrollo de la EK en epidemias en diferentes países, así como mayor incidencia en algunas regiones, es otro factor que sugiere una causa infecciosa. Otro elemento que soporta la etiología infecciosa es la ocurrencia de EK en hermanos de individuos afectados, los que se enferman la mayoría de las veces (54%) en los primeros 10 días del caso índice, lo que sugiere la exposición a un agente etiológico común, oponiéndose a una predisposición de tipo genético solamente. La baja frecuencia de la enfermedad en lactantes pequeños y en niños mayores y adultos, es consistente con la hipótesis de que la EK es causada por un agente al cual la mayoría de los adultos son inmunes y que los lactantes están protegidos por anticuerpos maternos. En contra de la etiología infecciosa, está el hecho de la escasa evidencia de que la EK pueda contagiarse de persona a persona, aunque la mayoría de los individuos podrían presentar una infección asintomática y solo algunos desarrollar síntomas de EK (1,14,24). Durante picos epidémicos de EK se ha tratado de hallar un factor ambiental común, describiéndose asociación con determinados limpiadores de alfombras, proximidad a lugares con agua estancada (lagunas, etcétera) y antecedente de una enfermedad respiratoria previa, pero ninguno ha sido probado (14). A pesar de una probable etiología infecciosa, no hay evidencia firme para ningún germen. Se ha vinculado con diferentes virus (parvovirus B19, citomegalovirus, virus de Epstein-Barr (25), otros herpes virus, virus del sarampión, etcétera), pero no se ha demostrado la implicancia como agente etiológico de ninguno de éstos. También se ha vinculado a la EK con bacterias (Mycoplasma pneumoniae, Propionibacterium acnes, Mycobacterium tuberculosis, Yersinia pseudotuberculosis, meningococo, etcétera) (26-28).
Existe una considerable evidencia clínica de una posible relación entre el síndrome de shock tóxico (SST) estafilocóccico y estreptocóccico con la EK. El SST presenta fiebre, afectación eritematosa de las mucosas, erupción cutánea con descamación, signos que comparte con la EK. En forma similar, las enfermedades estreptocóccicas mediadas por toxinas (escarlatina) presentan síntomas comunes con la EK. Además de los síntomas comunes al SST y la EK, se ha propuesto que ambas entidades comparten la patogenia del superantígeno, ya que ambas presentan severos disturbios inmunológicos, lo que se reafirma, además, por la respuesta favorable que estas dos entidades tienen al tratamiento con IGIV. Aunque no se ha podido establecer un vínculo definitivo, esta hipótesis es la que más aceptación presenta actualmente (1,14,20,23,24,29).
Patología
La EK es una vasculitis sistémica, que afecta fundamentalmente a las arterias de calibre mediano, aunque también están afectadas pequeñas y grandes arterias, capilares y venas. La afectación de las arterias coronarias es frecuente, siendo la más llamativa ya que puede producir infarto de miocardio. La EK fatal es indistinguible, desde el punto de vista anatomopatológico, de la poliarteritis nodosa, por lo que actualmente se acepta que representan la misma enfermedad. En la etapa aguda de la enfermedad se producen cambios inflamatorios en varios órganos (miocarditis, pericarditis, valvulitis, meningitis aséptica, neumonitis, linfadenopatía, hepatitis), siendo manifestaciones de la presencia de células inflamatorias en los tejidos comprometidos. En los vasos más afectados, como las arterias coronarias, se produce inflamación de la capa media, con edema y necrosis del músculo liso. Se pierde la integridad de la pared, lo que favorece la dilatación y formación de aneurismas. En etapas más avanzadas puede desarrollarse estenosis y oclusión arterial por trombosis sobreagregada. Aunque la vasculitis de las arterias coronarias es la más conocida, pueden afectarse otras arterias de gran y mediano tamaño con debilitamiento de la pared y formación de aneurismas y estenosis. Las mas comúnmente afectadas son: renales, ilíacas, paraováricas, paratesticulares, mesentéricas, pancreáticas, hepáticas, esplénicas y axilares (5,7,30).
Manifestaciones clínicas clásicas (3,6,7,9,10,23,24)
El diagnóstico de EK es clínico. No existe ningún examen ni test de laboratorio específico.
El diagnóstico se basa en el reconocimiento de los aspectos clínicos, que incluyen:
- fiebre de más de cinco días de duración;
- presencia de cuatro de cinco criterios principales (afectación ocular, cambios en labios y boca, erupción cutánea, cambios en las extremidades, adenopatía de cuello);
- sin otra causa que explique la enfermedad (tabla 1).
Los pacientes que presentan cuatro criterios principales (incluyendo la fiebre), pueden ser catalogados como EK, si presentan anomalías coronarias demostrables por ecocardiografía o coronariografía.
La bibliografía de origen japonés considera que si sólo se detectan cuatro de los síntomas clásicos, la enfermedad se cataloga como Kawasaki atípico, y si hay tres síntomas principales se cataloga como Kawasaki sospechoso. En Japón no se requiere estudio ecocardiográfico para realizar el diagnóstico cuando la sintomatología no es completa.
La bibliografía de origen estadounidense, en tanto, requiere para catalogar como Kawasaki atípico tres o menos de los síntomas clásicos y demostrar la presencia de aneurismas coronarios. El requisito de demostrar anomalías coronarias para catalogar un caso de EK como incompleto o atípico ha sido cuestionado por algunos autores, ya que se corre el riesgo de no diagnosticar la enfermedad en algunos pacientes (19,24).
En casos de fiebre de menos de cinco días de duración y presencia de los otros signos clásicos, puede realizarse diagnóstico de EK por parte de médicos experimentados en el diagnóstico de la enfermedad.
1) Fiebre: la fiebre es generalmente elevada: 39 a 40ºC, o mayor. El primer día de la fiebre es considerado el primer día de la enfermedad, aunque algunos pacientes pueden presentar uno o más de los otros síntomas clínicos el día antes del inicio de la fiebre. La duración de la fiebre, en ausencia de tratamiento, es de una a dos semanas, pero puede prolongarse hasta tres o cuatro semanas. Con el tratamiento adecuado (IGIV y AAS), la fiebre remite en uno a dos días después de iniciado el mismo.

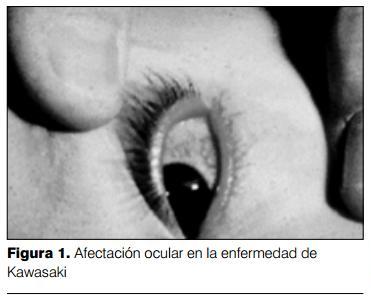
2) Afectación ocular: a nivel ocular se evidencia una hiperemia conjuntival, que es característica. La afectación de la conjuntiva bulbar es más evidente que la de la conjuntiva palpebral. No se acompaña de exudado, configurando una "conjuntivitis seca", aunque estrictamente no está afectada la conjuntiva, sino que es la presencia de pequeños vasos dilatados la que determina el enrojecimiento. La hiperemia conjuntival es más débil alrededor del iris, formando un halo más claro alrededor del mismo. Es frecuente la presencia de uveítis anterior, lo que puede detectarse por lámpara de hendidura (figura 1).
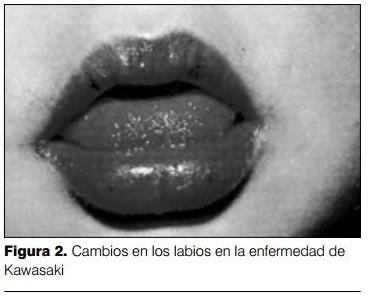
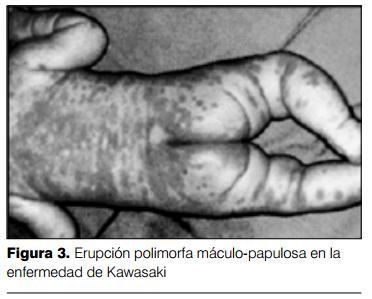
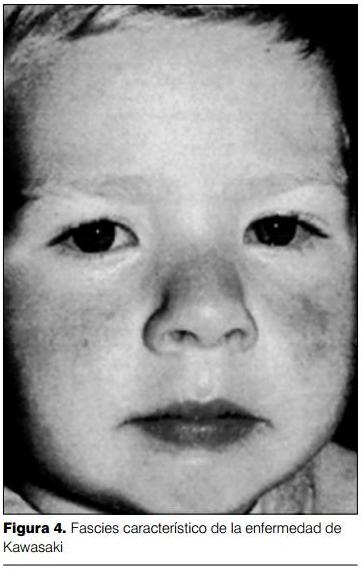
3) Cambios en labios y boca: los cambios en los labios se manifiestan por eritema, sequedad, fisuras y sangrado espontáneo o al mínimo tacto (figura 2). A nivel de la boca se evidencia eritema de la mucosa oral y faríngea, lengua aframbuesada con papilas prominentes eritematosas (figura 3). No se observan ulceraciones, exudados, ni manchas de Koplik. La lengua aframbuesada no es específica de la EK, y puede estar presente en otras enfermedades mediadas por toxinas (enfermedades estreptocóccicas y estafilocóccicas). Las alteraciones en ojos y boca determinan una imagen características (figura 4).
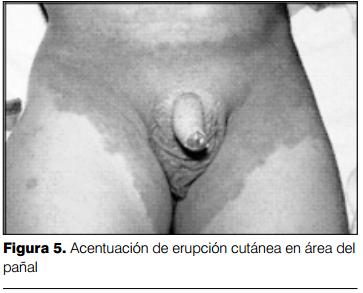
4) Erupción cutánea: las manifestaciones cutáneas pueden adoptar varias formas de presentación. Lo más característico es la presencia de un rash cutáneo eritematoso, polimorfo, máculo-papular no específico (figura 3). En ocasiones se presenta una erupción de tipo escarlatiniforme o con manifestaciones similares al eritema polimorfo. No se presentan vesículas ni bullas. En ocasiones pueden presentarse pequeñas pústulas, sobre todo en las zonas de extensión. Las lesiones elementales pueden acompañarse de púrpura, lo que es poco frecuente, pero característico. Las manifestaciones cutáneas se acentúan con la fiebre y son cambiantes. El eritema cutáneo es más marcado en el área del pañal, siendo fácilmente confundido con una dermatitis del pañal o candidiasis (figura 5). Las manifestaciones cutáneas en el área perineal también se presentan en enfermedades estreptocóccicas y estafilocóccicas.
La descamación cutánea en la fase aguda de la enfermedad está presente sobre todo en el área perineal, afectando el escroto en los varones y en las niñas los labios mayores (50%). Estas manifestaciones se observan tanto en los niños que utilizan pañales como en los que no los usan. Es frecuente la descamación perianal y la presencia de eritema a nivel del meato urinario.
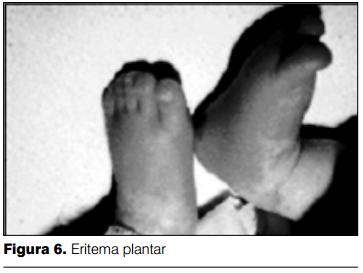
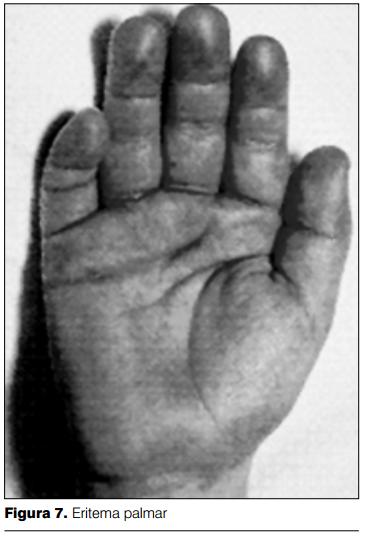
5) Cambios en las extremidades: a nivel de las extremidades se presenta eritema bilateral de las palmas y de las plantas (figuras 6 y 7). Se caracteriza por ser intenso, con cambios abruptos, llegando a desaparecer, dejando una piel de características normales, para posteriormente reaparecer. Se intensifica con la fiebre.
Las manos y pies presentan edema, sobre todo a nivel del dorso, que no deja godet. Es un edema "molesto", evidenciándose porque el niño "no quiere pararse", "no quiere caminar", o no puede sostener objetos con sus manos. Esto es debido al dolor determinado por la inflamación de pequeñas y medianas articulaciones. La inflamación articular puede afectar medianas y grandes articulaciones, aunque lo más frecuente es que afecte a las de pequeño tamaño, evidenciándose a nivel de las manos por la presencia de dedos de aspecto fusiforme. El dolor aumenta con la movilización, y en el lactante se manifiesta por llanto al movilizarlo.
En la etapa de convalecencia de la enfermedad, se observa descamación a nivel periungueal, que puede acompañarse de otras alteraciones en las uñas y descamación en las plantas. Después de uno a dos meses de la enfermedad, pueden desarrollarse surcos transversales en las uñas (líneas de Beau). Esta líneas desaparecen con el crecimiento de la uña y en ocasiones se produce el recambio de ésta.
6) Adenopatía cervical: la adenopatía cervical es el signo menos frecuente en comparación con las otras manifestaciones clínicas. Aunque se considera que el tamaño de la adenopatía debe tener como mínimo 1,5 cm, cuando está presente es notoria. Habitualmente es unilateral. Puede acompañarse de eritema de la piel, pero la adenopatía no presenta fluctuación, y no se obtiene pus si es puncionada. En ocasiones se han asistido niños con EK que inicialmente son catalogados como portadores de una adenitis de cuello, debiéndose tener en cuenta a la EK en los pacientes con adenopatía inflamatoria de cuello que no responde a los antibióticos y que no tiene otra causa que la explique.
Otras manifestaciones clínicas (3,6,7,9,10,23,24)
Una gran variedad de síntomas y signos se presentan en la EK, aunque no están incluidos en los criterios diagnósticos.
Un signo de gran valor, y que habitualmente está presente, es la irritabilidad. Ésta es intensa, a diferencia de otras enfermedades exantemáticas, y se presenta sobre todo en lactantes, lo que frecuentemente determina la realización de punción lumbar para descartar meningitis.
Aproximadamente un cuarto de los pacientes con EK tiene meningitis aséptica, con presencia de linfocitos en el líquido cefalorraquídeo, con recuento entre 15–100 elementos/mm3, con valores de glucosa normal y moderada elevación de las proteínas. No es necesaria la realización de punción lumbar en forma sistemática para el diagnóstico de EK.
Un signo de gran valor es la presencia de eritema e induración en el sitio de inoculación de la vacuna BCG, cuando ésta se ha aplicado en forma reciente (seis meses a un año). Este signo está presente en el 36% de los casos y ha sido incorporado en las guías diagnósticas de la EK en Japón (1).
Es frecuente la presencia de artralgia y de artritis. Se afectan las manos, rodillas y codos, y ocasionalmente caderas. La afectación articular puede estar presente en la primera semana de la enfermedad o puede presentarse en forma tardía, en la segunda o tercera semana de enfermedad. Desde que se realiza el tratamiento precoz, estas manifestaciones son menos frecuentes. El líquido sinovial, en las artritis precoces presenta elevado recuento de células (100.000 – 300.000 elementos/mm3), con predominio de polimorfonucleares, en tanto que las de manifestación tardía presentan menor celularidad (50.000 elementos/mm3), con 50% de polimorfonucleares.
Se presentan alteraciones a nivel hepático, manifestadas por elevación en el valor de las transaminasas, y en ocasiones se acompaña de ictericia.
La distensión de la vesícula biliar es frecuente, manifestándose por dolor en hipocondrio derecho. La ecografía evidencia una distensión alitiásica. No requiere tratamiento quirúrgico.
Otras manifestaciones menos frecuentes son diarrea, neumonitis, otitis media, urteritis con piuria estéril.
En ocasiones la EK puede presentarse con predominancia de síntomas de afectación del sistema nervioso central, como convulsiones, alteraciones del estado de conciencia, hemiplejia, parálisis facial, ataxia, sordera neurosensorial, infarto cerebral y derrame subdural. Algunos pacientes con manifestaciones neurológicas pueden quedar con secuelas permanentes (31,32,).
Las manifestaciones cutáneas pueden adoptar formas clínicas llamativas, como psoriasis guttata, erupciones verrugosas e hiperqueratósicas, con eliminación de moldes cutáneos de los dedos (33,34).
Manifestaciones cardíacas (3,6,7,9,10,23,24)
Las manifestaciones cardíacas son uno de los hechos más importantes en la EK.
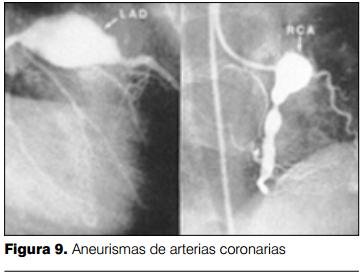
Dilatación y aneurismas coronarios
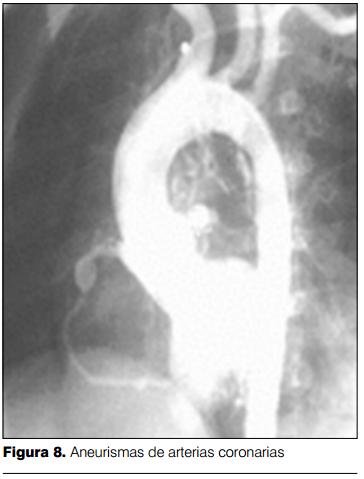
Aproximadamente el 15-25% de los niños no tratados desarrollan anomalías coronarias, incluyendo dilatación difusa y formación de aneurismas. La dilatación coronaria puede detectarse a partir de los 7-10 días de iniciada la enfermedad, pero es entre la tercera y cuarta semana en donde se produce el pico de mayor incidencia (35,36).
Es fundamental que la medida de las arterias coronarias sea realizada por ecocardiografista con amplia experiencia en niños.
El desarrollo de aneurismas coronarios se vincula con el riesgo de muerte súbita, por trombosis coronaria e infarto agudo de miocardio. En el seguimiento a largo plazo de los pacientes con dilataciones coronarias y aneurismas, se evidenció que el retroceso de las anomalías coronarias es un hecho frecuente, hasta en 50% de los casos, y en los restantes se evidenció disminución del tamaño de aneurismas, con o sin estenosis coronaria (25%), retroceso de aneurismas pero con estenosis coronaria (15%) y en los restantes finas irregularidades de los vasos coronarios pero sin estenosis (10%). En seguimientos a largo plazo, se estima que 40% de los pacientes con aneurismas coronarios persistentes y estenosis coronaria desarrollan infarto de miocardio (38).
La forma más severa de afectación coronaria es el desarrollo de aneurismas gigantes (³8mm). Estos aneurismas tienen menos probabilidad de retroceder y son frecuentes las complicaciones (trombosis, ruptura, estenosis). El porcentaje de niños con EK que desarrollarán aneurismas gigantes es variable, reportándose valores de entre 1 a 4%, con una mortalidad elevada (39).
Si se realiza tratamiento con IGIV y AAS antes de los 10 días de iniciada la enfermedad, la incidencia de alteraciones coronarias disminuye notoriamente, a porcentajes < de 5% (40).
La afectación de otras arterias puede ser causa de manifestaciones poco habituales aunque severas, como gangrena de extremidades por isquemia periférica, cuando se comprometen las arterias axilares o isquemia intestino-mesentérica por afectación de las arterias mesentéricas (41-44).
Infarto agudo de miocardio
Está vinculado a la presencia de anomalías coronarias. Su manifestación clínica es diferente a la del adulto. La forma de presentación es variada, inespecífica, y en ocasiones de diagnóstico difícil: shock, vómitos, dolor abdominal, palidez, diaforesis, llanto, debilidad. En niños grandes puede presentarse con dolor torácico. En la mayoría se presenta durante el sueño o el reposo y en ocasiones es asintomático. Puede ser causa de muerte súbita. Las manifestaciones electrocardiográficas y los cambios en las enzimas cardíacas son típicas. Es más frecuente en la fase subaguda de la enfermedad, momento de mayor frecuencia de aneurismas coronarios, pero también se puede presentar en la fase aguda o en etapas más tardías (35).
Otras manifestaciones cardíacas
La miocarditis es la manifestación cardíaca más frecuente en la fase aguda de la EK de compromiso no coronario, presentándose en hasta el 50% de los pacientes. Se manifiesta por taquicardia desproporcionada al grado de fiebre. No es frecuente que se acompañe de insuficiencia cardíaca congestiva o shock cardiogénico. Puede acompañarse de arritmias cardíacas. Los cambios electrocardiográficos que pueden evidenciar miocarditis son: prolongación del espacio PR, alteraciones en el segmento ST y onda T, y disminución de voltaje de la onda R La pericarditis con derrame pericárdico está presente en el 25% de los casos en la fase aguda. Puede presentarse compromiso valvular, evidenciado por valvulitis, sobre todo insuficiencia mitral (1).
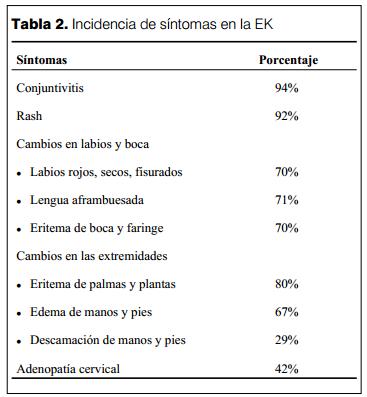
La frecuencia de cada de uno de los síntomas de la EK es variable (45) y se exponen en la tabla 2.
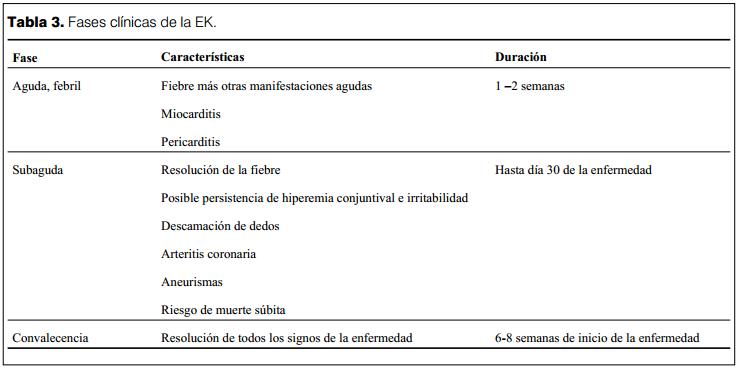
Fases clínicas de la enfermedad (3,6,7,9,10,23,24)
El curso de la EK puede ser dividido en tres fases clínicas: agudo, sub-agudo y convalecencia (tabla 3).
La fase aguda dura entre una a dos semanas, se caracteriza por fiebre y las manifestaciones clásicas de la enfermedad. Es en esta etapa que se presenta la miocarditis y la pericarditis. La arteritis está presente, pero rara vez se detectan la presencia de aneurismas por ecocardiografía.
La fase subaguda comienza cuando retrocede la fiebre, la erupción cutánea y la adenopatía cervical, pero puede persistir irritabilidad, anorexia y la hiperemia conjuntival, con descamación a nivel de los dedos y pies. Esta fase se prolonga habitualmente hasta cuatro semanas de iniciada la enfermedad. Los aneurismas de las arterias coronarias se desarrollan en esta fase y el riesgo de muerte súbita es elevado.
La etapa de convalecencia se prolonga hasta que retrocede toda la sintomatología y finaliza cuando se normaliza el valor de la velocidad de eritrosedimentación (VES), habitualmente en plazos de 6 a 8 semanas de iniciada la enfermedad.
"Kawasaki atípico"
Desde la perspectiva clínica, la necesidad de reconocer tempranamente la EK ha permitido definir, cada vez más, que un número importante de casos de la enfermedad no reúne las características clásicas y cerca de un 40% de los pacientes que desarrollan alteraciones coronarias tienen presentaciones atípicas (20).
Estos niños presentan fiebre y menos de los cuatro síntomas requeridos para diagnosticar EK. A estos casos se les denomina "Kawasaki atípico" o "Kawasaki incompleto". Se estima que al menos 10% de los casos de EK son incompletos (24). Estos casos son más frecuentes en niños pequeños, sobre todo lactantes, en donde la probabilidad de desarrollar aneurismas coronarios es mayor. El diagnóstico en estas situaciones puede ser difícil y se debe tener un alto índice de sospecha ante niños de corta edad que presentan fiebre prolongada sin una causa evidente, o con algunas de las manifestaciones clásicas de la enfermedad. El estudio ecocardiográfico puede ser de gran ayuda, si detecta la presencia de aneurismas coronarios, pero su ausencia no invalida sospechar la enfermedad. Se ha vinculado una mayor incidencia de aneurismas coronarios en los pacientes con Kawasaki atípico, probablemente por demoras en el diagnóstico (7,23). En revisiones recientes se ha determinado que cada vez se diagnostican con mayor frecuencia casos de EK que no cumplen con los criterios diagnósticos clásicos, y que reciben tratamiento con IGIV más AAS. Esta conducta parece estar justificada, ya que esperar a realizar el diagnóstico de EK sobre la base de los criterios diagnósticos clásicos, determinaría mayor incidencia de desarrollo de aneurismas coronarios, al excluir a los niños que se presentan con síntomas incompletos (46). El reconocimiento y tratamiento temprano de la enfermedad son vitales para prevenir las complicaciones cardíacas (47). Los pacientes con Kawasaki atípico presentan las mismas alteraciones de laboratorio que los que se presentan con la forma clásica y completa.
Del análisis de los datos epidemiológicos mundiales, surge, además, la preocupación sobre el retardo en la formulación del diagnóstico y del tratamiento en los niños mayores de cinco años y, en especial, en los mayores de 10 años (20).
Diagnóstico diferencial
El diagnóstico diferencial se plantea con enfermedades febriles que cursan con erupción cutánea: escarlatina, síndrome de piel escaldada, síndrome de shock tóxico, síndrome de Stevens-Johnson, reacción a drogas, artritis reumatoidea juvenil, infecciones virales (adenovirus), sarampión, leptospirosis, infección por ricketsias. Habitualmente estas entidades pueden diferenciarse por medio de la clínica.
La infección viral por adenovirus puede tener presentación clínica similar a EK. La detección de antígenos virales por medio de tests rápidos (inmunofluorescencia) en aspirado nasofaríngeo de secreciones puede ayudar al diagnóstico (48).
Cuando se plantea diagnóstico diferencial con escarlatina, el aislamiento de estreptococo beta hemolítico de grupo A (EBHGA) en el exudado faríngeo puede ayudar al diagnóstico. Se debe tener en cuenta que 20-25% de los niños pueden ser portadores de EBHGA, por lo que algunos niños con EK pueden tener cultivos positivos por este organismo. Como los pacientes con escarlatina tienen una rápida mejoría cuando se tratan con antibióticos, se puede diferenciar estas entidades en función de la respuesta clínica rápida, luego de 24 a 48 horas de tratamiento (1).
La presentación de un paciente en la emergencia con "ojo rojo", fiebre prolongada y erupción cutánea, puede ser debida a diferentes causas (45). Tener en cuenta las diferentes etiologías puede ayudar al diagnóstico en casos difíciles. El análisis ocular con lámpara de hendidura puede evidenciar uveítis anterior (tabla 4).
En los niños mayores de cinco años y adolescentes puede equivocarse el diagnóstico, por no tener en mente a la EK en este grupo etario (20,49).
En casos de EK que no responden al tratamiento con IGIV, debe tenerse en cuenta la poliarteritis nodosa infantil, artritis reumatoidea juvenil de inicio sistémico y enfermedades malignas como el linfoma (50).
Laboratorio
Los hallazgos de laboratorio en EK no son específicos de la enfermedad, pero presentan características particulares.
En la fase aguda de la enfermedad, existe aumento de los glóbulos blancos con predominio de neutrófilos con desviación a izquierda de la fórmula leucocitaria. Puede presentarse eosinofilia, hasta en un tercio de los casos (51). La leucopenia no se presenta en la EK y su presencia debe hacer dudar del diagnóstico (7). Es frecuente la anemia normocítica y normocrómica. Se ha vinculado con aumento de lesiones coronarias, sobre todo si es intensa (23). El recuento plaquetario es normal en la fase aguda de EK, presentándose trombocitosis en la fase sub-aguda (segunda a tercera semana), elemento característico de la EK, con valores elevados, de hasta 1 millón/dl. La presencia de plaquetopenia en la fase aguda se asocia con mayor incidencia de lesiones coronarias e infarto de miocardio. Es una manifestación poco frecuente, invocándose un mecanismo autoinmune ya que responde al tratamiento con IGIV (52). La VES y la PCR están elevadas en la fase aguda y pueden persistir valores elevados durante cuatro a seis semanas. La persistencia de VES elevada después que la fiebre ha desaparecido puede ayudar a distinguir la EK de otras enfermedades febriles acompañadas de erupción cutánea (45).
La evolución de los valores de la VES y PCR no necesariamente van juntas, ya que elevaciones modestas de la VES pueden acompañarse de valores muy elevados de la PCR (45).
En etapas precoces de EK existe hipercoagulabilidad asociada a un aumento del "turnover" plaquetario y a depleción del sistema fibrinolítico (45).
En el examen de orina se detecta piuria estéril. Las células se originan en la uretra por lo que no son detectadas si la orina se recoge de la vejiga por cateterismo vesical. No son detectadas por las tirillas reactivas de uso habitual (45).
A nivel del líquido cefalorraquídeo se detecta en 1/3 de los pacientes con EK a los que se les realizó punción lumbar, pleocitosis con predominio de monoculeares. No es frecuente la hipoglucorraquia ni aumento de proteínas (53).
Es frecuente el aumento en el valor de las transaminasas en la fase aguda, que pueden adoptar un perfil de tipo colestático con aumento de las bilirrubinas. La GGT está elevada en 2/3 de los casos. La presencia de hipoalbuminemia se vincula a mal pronóstico (21).
Son frecuentes las alteraciones inmunológicas. En las etapas iniciales existe disminución de IgG, y en la fase subaguda aumento en valores de IgG, IgM, IgA e IgE (7,23).
En la evaluación de un paciente con EK con dificultades para catalogarlo como tal, es frecuente la realización de otros exámenes para descartar otros diagnósticos: hemocultivos, AELO, exudado faríngeo, perfil de autoinmunidad (anticuerpos antinucleares, factor reumatoideo, anticuerpos ANCA), serología para bacterias y virus.
La evaluación cardiológica requiere radiografía de tórax, ECG y ecocardiografía. La dosificación de troponina en la fase aguda de EK, puede indicar presencia de miocarditis (54), aunque no es un hecho consistente (55) por lo que no es de uso rutinario.
Es un error conceptual esperar signos de laboratorio como condicionante del diagnóstico (56).
Tratamiento (7,14,17,23,24,40,45)
El tratamiento estándar de EK se realiza con altas dosis de IGIV y AAS.
Inmunoglobulina de uso intravenoso (IGIV)
La administración de IGIV 2 gr/kg en infusión de 12 horas, antes del 10º día de iniciada la enfermedad, reduce la incidencia de aneurismas coronarios de 20% a menos de 4%. Produce una rápida defervescencia de la fiebre y los síntomas asociados con rápida normalización de los valores de los reactantes de fase aguda, y además mejora la función miocárdica. En los pacientes con falla cardíaca aguda puede ser necesaria la administración en forma más lenta para evitar sobrecarga de volumen.
Ácido acetilsalicílico (AAS)
El AAS se inicia con dosis elevadas, de 80-100 mg/kg/día divididos en 4 dosis diarias, buscando un efecto antiinflamatorio, que se mantiene hasta por lo menos pasados 3-4 días de apirexia (algunos autores recomiendan mantener esta dosis hasta el día 14 de iniciada la enfermedad). Ésta es la dosis recomendada por la AAP y la AHA.
En Japón se utilizan dosis más bajas de AAS (30-50 mg/kg/día).
Posteriormente se continúa con dosis menor, 3-5 mg/kg/día en una toma diaria, buscando un efecto antiplaquetario y antitrombótico, que se mantendrá como mínimo seis semanas. Si se detectan anomalías coronarias se mantiene el tratamiento profiláctico con AAS, hasta que se resuelva el problema.
En EK está alterada la farmacocinética del AAS, con disminución de la absorción y aumento del clearence, por lo que es difícil mantener rangos de salicilemia en valores terapéuticos. Habitualmente el tratamiento con AAS en EK no determina mayor riesgo de intoxicación, pero es recomendable monitorizar los niveles séricos, sobre todo cuando se utilizan dosis elevadas.

En los pacientes en los que se demoró el inicio del tratamiento más allá del décimo día de la enfermedad, está indicado el tratamiento con IGIV si persisten febriles o presentan signos de actividad de la enfermedad, ya que hay mayor posibilidad de desarrollar aneurismas coronarios. Si el paciente se presenta con más de 10 días de evolución, con varios días de apirexia y descamación, no está indicada la administración de IGIV, ya que no hay datos que sugieran la eficacia en esta situación, ya que la IGIV no previene la enfermedad coronaria después de la fase aguda de la enfermedad cuando la respuesta inflamatoria ha disminuido.
Si se realiza el diagnóstico de EK en forma precoz, antes del quinto día de iniciada la enfermedad, está justificado el tratamiento con IGIV, ya que se ha determinado que mejora el pronóstico a largo plazo de secuelas coronarias y disminución de la duración de los síntomas (10,57). Otros autores no han confirmado estos resultados, incluso informan mayor incidencia de lesiones coronarias cuando se realiza tratamiento temprano (tercer día), con la limitante que no utilizan el régimen de 2 g/kg de IGIV sino que utilizan el régimen de 400 mg/kg/día en tres a cuatro días (el régimen de 2 g/kg no está aprobado en Japón en forma sistemática por razones económicas, aunque ha demostrado ser superior que el de dosis menores) (58).
En los pacientes que no cumplen con los criterios clásicos, "Kawasaki atípico" o "Kawasaki incompleto", no debe retrasarse el tratamiento (14). El médico actuante debe ejercer su mejor juicio clínico para identificar y tratar a los pacientes con EK que no cumplen con los requisitos clásicos de caso definitivo, para reducir la incidencia de complicaciones cardíacas y no ceñirse estrictamente a los plazos establecidos. El requisito de fiebre de cinco días o más no debe entenderse literalmente. La mención de T. Kawasaki de 5 días o más fue hecha en otro contexto, en el que el sarampión era un problema diagnóstico diferencial (58).
Falla de tratamiento
No todos los pacientes con EK responden al tratamiento inicial con IGIV y persisten febriles después de su administración o reiteran fiebre y otros síntomas de la enfermedad en los primeros dos días luego de un período de apirexia. Se estima que 10% a 30% de los pacientes no responden al tratamiento inicial con IGIV. El tratamiento óptimo de estos pacientes no está definitivamente establecido, pero actualmente existe consenso en reiterar la IGIV en caso de falla de tratamiento. En algunas series de pacientes se han requerido reiterar dos a tres veces las infusiones de IGIV (11,59-64).
Corticoides
Muchos clínicos proponen, en caso de falla de tratamiento, asociar a la IGIV el uso de corticoides. Aunque las referencias iniciales sobre los corticoides vinculaban su uso a mayor incidencia de lesiones coronarias, actualmente se plantea utilizarlos en aquellos pacientes que no responden a la primera o segunda infusión de IGIV, obteniéndose resultados favorables. Se utiliza metilprednisolona a dosis de 30 mg/kg/día durante tres días (11,59-64).
En casos seleccionados de pacientes que no responden a estos regímenes terapéuticos, se han utilizado inmunosupresores como la ciclosporina (11,59-64). Comunicaciones recientes, que hacen hincapié en el mecanismo lesional a nivel arterial mediado por radicales libres de oxígeno, proponen el uso de sustancias antioxidantes (alfa-tocoferol, ácido ascórbico), en caso de no respuesta al tratamiento inicial (65). Más estudios se necesitan para este grupo de pacientes con la finalidad de establecer protocolos de tratamiento ampliamente probados.
Si se desarrollan aneurismas coronarios debe continuarse el tratamiento con AAS en tanto éstos persistan. Algunos autores proponen sustituir el AAS o agregar un antiagregante plaquetario como el dipiridamol a dosis de 2-3 mg/kg dos a tres veces por día.
Si se desarrolla infarto agudo de miocardio, es necesario realizar tratamiento trombolítico.
En caso de gangrena de extremidades, es necesario un tratamiento complejo: mejora de la función cardíaca (inotrópicos), vasodilatadores (nitroprusiato, prostaglandinas, bloqueo caudal) para el vasoespasmo y agentes antitrombóticos (heparina, warfarina, estreptoquinasa, dipiridamol, AAS a bajas dosis), con necesidad de participación de equipo multidisciplinario (cardiólogo, cirujano vascular, cirujano plástico, intensivista) para evitar amputaciones.
Factores de riesgo
Actualmente se otorga gran importancia a la detección de factores de riesgo que pronostiquen falla de tratamiento o mayor incidencia de lesiones coronarias, para de esta manera estar más vigilantes en este grupo de pacientes.
Los hallazgos de laboratorio traducen la intensidad de la respuesta inflamatoria. Los valores elevados de leucocitos con predominio de neutrófilos (menos de 68%), el descenso en el valor de la hemoglobina (más de 10 g/dl), la hipoalbuminemia, la plaquetopenia, aumento de enzimas hepáticas, sobre todo LDH mayor a 590 UI/l, valores elevados de la PCR y VES, persistencia de leucocitosis elevada después de IGIV, presencia de factor estimulante de colonias de granulocitos, se han vinculado con mal pronóstico.
El score de Harada se utiliza para predecir lesiones en las arterias coronarias en pacientes con EK, si presenta al menos cuatro de los siguientes siete elementos: 1) leucocitosis mayor a 12.000 elementos/mm3, 2) recuento plaquetario menor de 35 x 104 elementos/mm3, 3) PCR elevada, 4) hematocrito menor a 35%, 5) albúmina menor a 3,5 g/dl, 6) edad mayor de 12 meses y 7) sexo masculino (66-71).
Otro factor de riesgo de desarrollar lesiones coronarias es la recurrencia de la enfermedad. La frecuencia de casos recurrentes es de 3% (72,73).
Manejo después de la fase aguda
Si el paciente tiene una respuesta favorable al tratamiento con IGIV + AAS, habitualmente se produce remisión de los síntomas en 24 horas. Debe mantenerse la internación durante 24-48 horas después de lograda la apirexia. Los controles clínicos deben realizarse en forma semanal durante la fase subaguda y de convalecencia. Es necesario reiterar un ecocardiograma a las 2-3 semanas del inicio de la enfermedad, que debe reiterarse a las 6-8 semanas, si el estudio previo fue normal. Debe realizarse ecocardiograma al año de la enfermedad, y si éste es normal no se recomienda realizar nuevos estudios. Los exámenes de laboratorio (hemograma, VES, PCR) se repiten en los mismos plazos.
Cuando los valores de los reactantes de inflamación se normalizan (habitualmente a las 6-8 semanas) y si el ecocardiograma no demuestra alteraciones coronarias, se suspende la administración de AAS.
Los plazos señalados para los controles pueden adaptarse a situaciones particulares, en función de la presencia de factores de riesgo previamente establecidos.
La administración de vacunas con virus vivos atenuados (sarampión, rubéola, paperas, varicela) debe posponerse entre 6 a 11 meses después de haber recibido IGIV, ya que ésta puede impedir el desarrollo de una respuesta inmune adecuada.
En caso de exposición al virus de varicela en un paciente con EK que no tuvo la enfermedad y que está recibiendo AAS, debe considerarse la suspensión de AAS, por riesgo de desarrollar síndrome de Reye. En este caso puede sustituirse el AAS por dipiridamol.
También debe considerarse la administración de vacuna antigripal a los pacientes que permanecen en tratamiento crónico con AAS, para reducir el riesgo de síndrome de Reye.
Los niños que presentan anomalías coronarias deben controlarse con cardiólogo pediatra, quien decidirá la frecuencia de la realización de estudios (ECG, ecocardiograma, estudios dinámicos, coronariografía), necesidad de tratamiento anticoagulante y tratamientos quirúrgicos.
Si no se detectan anomalías coronarias en la fase aguda o subaguda de la enfermedad, no hay evidencia firme de alteraciones vasculares a largo plazo y los pacientes no presentan mayor mortalidad que el resto de la población (74), aunque actualmente este concepto está en revisión.
Se ha establecido que los pacientes que padecieron EK presentan alteraciones electrocardiográficas con mayor frecuencia (tres veces) que la población control (75), alteraciones isquémicas silentes cuando se estudian con test de stress con dobutamina (76) o perfiles lipídicos alterados con potencial riesgo de aterosclerosis. Más estudios se requieren para sacar conclusiones definitivas.
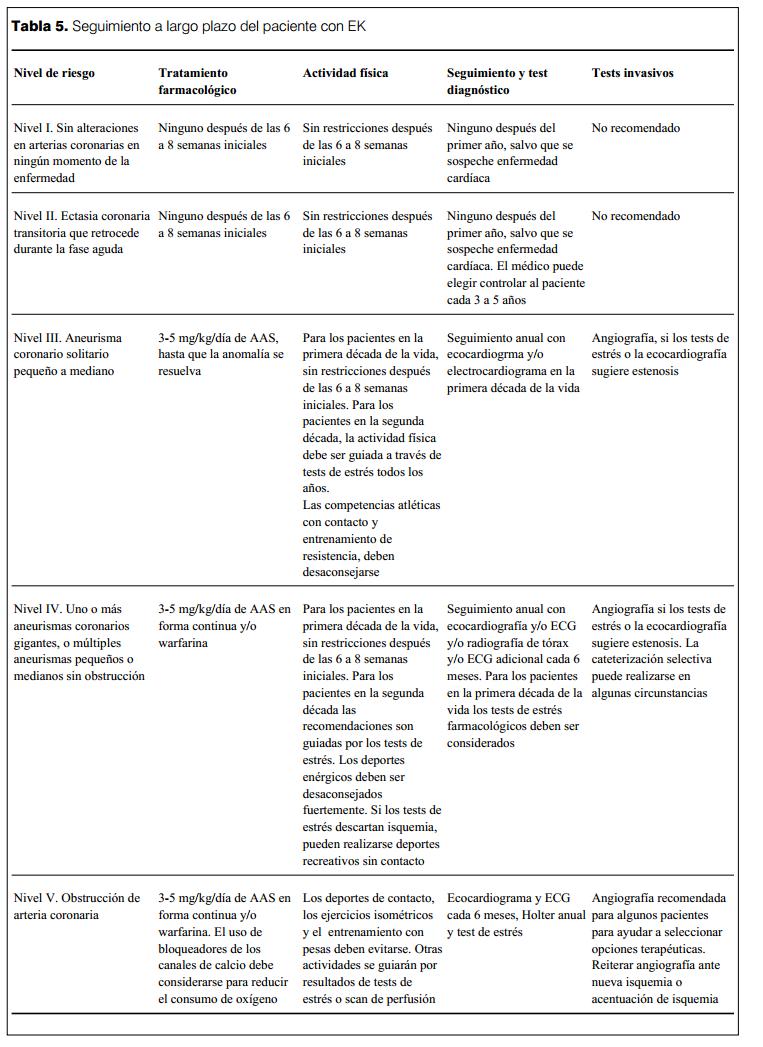
Manejo a largo plazo
El manejo a largo plazo de los pacientes con EK depende del grado de afectación de las arterias coronarias y se realiza en base a la estratificación por grupos según el riesgo relativo de isquemia miocárdica. La AHA propone el esquema de seguimiento que se ve en la tabla 5 (77). El seguimiento por especialistas requiere el acceso a tecnología de estudio avanzada como: ergometría de esfuerzo, tests de estrés farmacológicos (dobutamina, dipiridamol, adenosina) para niños pequeños que no pueden realizar esfuerzo físico en bicicleta o en la cinta, estudios radioisotópicos de perfusión miocárdica, valoración con ecocardiografía de la motilidad regional de la pared ventricular, utilización de resonancia nuclear magnética, etcétera.
Key words: MUCOCUTANEOUS LYMPH NODE
SYNDROME
Bibliografía
1. Burns JC, Kushner HI, Bastian JF, Sjike H, Shimizu C, Matsubara T, et al. Kawasaki Disease: a brief history. Pediatrics 2000; 106(2): 27.
2. Kawasaki T. Pediatric acute mucocutaneous lymph node syndrome: clinical observation of 50 cases. Jpn J Allergy 1967; 16: 178-222.
3. Yamamoto T, Oya T, Watanabe A. Clinical features of Kawasaki disease. Jpn J Pediatr 1968; 21: 291-7.
4. Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 1974; 54(3): 271-6.
5. Landing BH, Larson EJ. Are infantile periarteritis nodosa with coronary artery involvement and fatal mucocutaneous lymph node syndrome the same? Comparison of 20 patients from North America with patients from Hawaii and Japan. Pediatrics 1976; 59: 651-62.
6. Melish ME, Hicks RM, Larson EJ. Mucocutaneous lymph node syndrome in the United States. Am J Dis Child 1976; 130: 599-607.
7. Rowlay AH, Shulman ST. Kawasaki Syndrome. Clin Microbiol Rev 1998; 11(3): 405-14.
8. Morens DM, O´Brien RJ. Kawasaki disease in the United States. J Infect Dis 1978; 137: 91-3.
9. American Academy of Pediatrics. Kawasaki Disease. In: Peter G, ed. 1988: Red Book: Report of the Committee on Infectious Diseases. 21th. ed. Elk Grove Village: American Academy of Pediatrics, 1988: 251-4.
10. Dajani AS, Taubert KA, Gerber MA, Shulman ST, Ferrieri P, Freed M, et al. Diagnosis and treatment of Kawasaki disease in children. Circulation 1993; 87: 1776-80.
11. Wright DA, Newburger JW, Baker A, Sundel RP. Treatment of immune globulin-resistant Kawasaki disease with pulsed doses of corticosteroids. J Pediatr 1996; 128: 146-149.
12. Furukawa S, Matsubara T, Umezawa Y, Motohashi T, Ino T, Yabuta K. Pentoxifilline and intravenous gamma globulin combination therapy for acute Kawasaki disease. Eur J Pediatr 1994; 153: 663-7.
13. Caggiani M, Rubio I, Parada P, Lagomarsino G, Guidobono G, Mañé Garzón F. Enfermedad de Kawasaki. A propósito de un caso. Arch Pediatr Urug 1989: 60(1-4): 65-8.
14. Laupland KB, Dele Davies H. Epidemiology, Etiology, and Management of Kawasaki Disease: State of the art. Pediatr Cardiol 1999; 20: 177-83.
15. Stanley TV, Grimwood K. Clasical Kawasaki disease in a neonate. Arch Dis Chil Fetal Neonatol 2000; 86: F135-F136.
16. Stocheim JA, Innocentini N, Shulman S. Kawasaki disease in older children and adolescents. J Pediatr 2000; 137(2): 250-2.
17. McCrindel BW, Shulman ST, Burns JC, Kato H, Gersony WM, Newburger JW. Meeting Report. Summary and abstracts of the Sixth International Kawasaki Disease Symposium. Pediatr Res 2000; 47(4): 544-8.
18. Du Z, Zhang T, Liang L, Meng, X, Li T, Kawasaki T, et al. Epidemiologic picture of Kawasaki disease in Beijing from 1995 through 1999. Pediatr Infect Dis J 2002; 21: 103-7.
19. Royle JA, Wiliams K, Elliot E, Choller G, Nolan T, Allen R, et al. Kawasaki disease in Australia 1993-95. Arch Dis Child 1998; 78: 33-9.
20. Banfi A. Enfermedad de Kawasaki. Rev Chil Pediatr 2001; 76(6): 487-95.
21. Yanagawa H, Yashiro M, Nakamura Y, Kawasaki T, Kato H. Epidemiological pictures of Kawasaki disease in Japan; form the nationwide incidence survey in 1991 and 1992. Pediatrics 1995; 149: 779-83.
22. Dean AG, Melish ME, Hicks R, Palumbo E. An epidemic of Kawasaki syndrome in Hawaii. J Pediatr 1982; 100: 552-7.
23. Rowley AH, Shulman ST. Síndrome de Kawasaki. Clin Ped NA 1999; 2: 341-59.
24. Kawasaki T. Kawasaki Disease. Acta Paediatr 1995; 84: 713-5.
25. Machado K, Gutiérrez S, Pirez C. Enfermedad de Kawasaki asociada a virus de Epstein-Barr. Arch Pediatr Urug 2002; 73(4): 220-5.
26. Leen C, Ling S. Mycoplasma infection and Kawasaki disease. Arch Dis Child 1996; 75: 226-7.
27. Ramanan AV, Baildam EM. Kawasaki disease following meningococcal septicaemia. Arch Dis Chil 2002; 87: 170.
28. Shibata M, Ezaki T, Hori M, Nagashima M, Morishima T. Isolation of a Kawasaki disease-associated bacteria sequence from peripheral blood leukocytes. Pediatrics International 1999; 41: 467-473.
29. Leung D, Meissner HC, Shulman ST, Mason WH, Gerber MA, Glode MP, et al. Prevalence of superantigen-secreting bacteria in patients with Kawasaki disease. J Pediatr 2002; 104(6): 742-6.
30. Foster BJ, Bernard C, Drummond K. Kawasaki disease complicated by renal artery stenosis. Arch Dis Child 2000; 83: 253-5.
31. Tabakari B, Mahdhaoui A, Selmi H, Ytacoub M, Essoussi A. Kawasaki disease with predominant Central Nervous System involvement. Pediatr Neurol 2001; 25(3): 239-41.
32. King WJ, Schlieper A, Birdi N, Capelli M, Korncluk Y, Rowe P. The effect of Kawasaki disease on cognition and behavior. Arch Pediatr Adolesc Med 2000; 154: 463-8.
33. Garty B, Mosseri R, Finkelstein Y. Guttate Psoriasis following Kawasaki disease. Pediatr Dermatol 2001; 18(6): 507-8.
34. Passeron T, Olivier V, Sirvent N, Khalfi A, Boutte P, Lacour JP. Kawasaki disease with exceptional cutaneous manifestations. Eur J Pediatr 2002; 161: 228-30.
35. Kato H, Hichinose E, Kawasaki T. Myocardial infarction in Kawasaki disease: clinical analyses in 195 cases. J Pediatr 1986; 108: 923-7.
36. González E, Villanueva J, Ros J, Pons M, Ruiz S. Enfermedad de Kawasaki. Presentación de cincuenta casos. An Esp Pediatr 1999; 50(1): 39-43.
37. de Zorzi A, Colan SD, Gauvreau K, Baker AL, Newburger JW. Coronary artery dimensions may be misclassified as normal in Kawasaki disease. J Pediatr 1998; 133: 254-8.
38. Kato H, Inoue O, Akagi T, Sato N, Hashimo K, Maeno Y, et al. Long-Term consequences of Kawasaki disease. Circulation 1996; 94: 1279-85.
39. Nakamura Y, Yashiro M, Tanihara S, Ojima T, Yanagawa H. Giant coronary aneurysms due to Kawasaki disease: a case-control study. Pediatrics International 2002; 44: 254-8.
40. Newburger JW, Takahashi M, Beiser AS, Burns JC, Bastian J et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med 1991; 324: 1633-9.
41. Bachiri A, Francart C, Godart F, Breviere GM, Vaskman G, Martinot V, et al. Ischemie de la main revelant une maladie de Kawasaki. Arch Pediatr 2000; 7: 1307-10.
42. Chang JS, Lin JS, Peng CT, Tsai CH. Kawasaki disease complicated by peripheral gangrene. Pediatr Cardiol 1999; 20: 139-42.
43. Beiler HA, Schmidt KG, von Herbay A, Loffler W, Daum R. Ischemic small bowel strictures in a case of incomplete Kawasaki disease. J Pediatr Surg 2001; 36(4): 648-50.
44. Krohn C, Till H, Haraida S, Kurnik K, Bohem R, Grantzow R, et al. Multiple intestinal stenoses and peripheral gangrene: a combination of two rare surgical complications in a child with Kawasaki disease. J Pediatr Surg 2001; 36(4): 651-3.
45. Burns JC. University of California, San Diego. Kawasaki Disease Program. www.pediatrics.ucsd.edu/divisions/Inmunology-Allergy/outline.asp
46. Witt MT, Minich LL, Bohnsack JF, Young PC. Kawasaki disease: more patients are being diagnosed who do not meet American Heart Association criteria. Pediatrics 1999; 104(1): www.pediatrics.org/cgi/content/full/104/1/e10.5 screens.
47. Curtis N. Kawasaki Disease. Early recognition is vital to prevent cardiac complications. Editorial. BMJ 1997; 315: 322-3.
48. Barone SR, Pontrelli LR, Krilov LR. The differentiation of Classic Kawasaki Disease, Atypical Kawasaki Disease, and acute adenoviral infection. Use of clinical features and a rapid direct fluorescent antigen test. Arch Pediatr Adolesc Med; 2000: 154(5): 453-6.
49. Zamora ME, Arana I, Bertó J, Zapatero M, Megías A. Enfermedad de Kawasaki de presentación incompleta. Acta Pediatr Esp 2000; 58: 467-9.
50. Brogan PA, Bose A, Burgner D, Shingadia D, Tulloh R, Michie C, et al. Kawasaki disease: an evidence based approach to diagnosis, treatment and proposals for future research. Arch Dis Child 2002; 86: 286-90.
51. Terai M, Yasukawa K, Honda T, Jibiki T, Hirano K, Sato J, et al. Peripheral blood esosinophilia and eosinophil accumulation in coronary microvessels in acute Kawasaki disease. Pediatr Infect Dis J 2002; 21: 777-80.
52. Venglarick JS, Mouhab A. Severe thrombocytopenia as a presenting manifestation of Kawasaki disease. Arch Pediatr Adolesc Med 1995; 149: 215-216.
53. Dengler L, Capparelli E, Bastian J, Bradley D, Glode M, Santa S, et al. Cerebrospinal fluid profile in patients with acute Kawasaki disease. Pediatr Infect Dis J 1998; 17(6): 478-81.
54. Kim M, Kim K. Elevation of cardiac troponin I in the acute stage of Kawasaki disease. Pediatr Cardiol 1999; 20: 184-8.
55. Checchia P, Borensztajn J, Shulman S. Circulating cardiac Troponin I levels in Kawasaki disease. Pediatr Cardiol 2001; 22: 102-6.
56. Schonhaut L, Herrera P, Acevedo K, Alvarez P. Enfermedad de Kawasaki en el Hospital del Río: análisis clínico epidemiológico. Rev Chil Pediatr 2001; 72(4): 319-27.
57. Tse S, Silverman E, McCrindle B, Yeung R. Early treatment with intravenous inmunoglubullin in patients with Kawasaki disease.J Pediatr 2002; 140(4).450-5.
58. Nomura Y, Masuda K, Yoshinaga M, Semeshima K, Miyata K. Patients diagnosed with Kawasaki disease before the fifth day of illness have a higher risk of coronary artery aneurysm. Pediatrics International 2002; 44: 353-7.
59. Raman V, Kim J, Sharkey A, Chatila T. Response of refractory Kawasaki disease to pulse steroid and cyclosporin therapy. Pediatr Infect Dis J 2001; 6: 635-7.
60. Burns J, Capparelli E, Brown J, Newburger JW, Glode M. US/Canadian Kawasaki syndrome study group. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. Pediatr Infect Dis J 1998; 17(12): 1144-8.
61. Wallace C, French J, Kahn S, Sherry D. initial Intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics 2000; 105(6): www.pediatrics.org/cgi/content/full/105/6/e78 4 screens.
62. Han R, Silverman E, Newman A, McCrindel B. Management and outcome of persistent or recurrent fever after initial intravenous gamma globulin therapy in acute Kawasaki disease. Arch Pediatr Adolesc Med 2000; 154: 694-9.
63. Shinohara M, Sone K, Tomamasa T, Morikawa A. Corticosteroids in the treatment of the acute phase of Kawasaki disease. J Pediatr 1999; 135(4): 465-9.
64. Newburger JW. Treatment of Kawasaki disease: Corticosteroids revisited. J Pediatr 1999; 135: 411-3.
65. Shen C, Wang N. Antioxidants may mitigate the deterioration of coronary arteritis in patients with Kawasaki disease unresponsive to high-dose intravenous gamma-globulin. Pediatr Cardiol 201; 22: 419-22.
66. Newburger JW. Kawasaki disease: Who is at risk?. J Pediatr 2000; 137: 149-52.
67. Oki I, Tanihara S, Nakamura Y, Yanagawa H. A multicenter collaborative study on the risk factors of cardiac sequelae due to Kawasaki disease: a one-year follow-up study. Acta Paediatr 2000; 89: 1435-8.
68. Fukinishi M, Kikkawa M, Hamana K, Onedara T, Matsuzaki Y, Hara J. Prediction of non-responsiveness to intravenous high-dose gamma-globulin therapy in patients with Kawasaki disease at onset. J Pediatr 2000; 127(2): 172-6.
69. Silva A, Maeno Y, Hashmi A, Smallborn J, Silverman E, McCrindle B. Cardiovascular risk factors after Kawasaki disease: A case-control study. J Pediatr 2001; 138(3): 400-5.
70. Samada K, Igarashi H, Shiraishi H, Hatake K, Momoi M. Increased serum granulocyte colony-stimulating factor correlates with coronary dilatation in Kawasaki disease. Eur J Pediatr 2002; 161: 538-41.
71. Zhang T, Yanagawa H, Nakamura Y. Factors relating to the cardiac sequelae of Kawasaki disease one month after initial onset. Acta Paediatr 2002; 91: 517-20.
72. Nakamura Y, Yanagawa H, Ojima T, Kawasaki T, Kato H. Cardiac sequelae of Kawasaki disease among recurrent cases. Arch Dis Child 1998; 78: 163-5.
73. Nakamura Y, Oki I, Tanihara S, Ojima T, Yanagawa H. Cardiac sequelae in recurrent cases of Kawasaki disease: A comparison between the initial episode of the disease and a recurrence in the same patients. Pediatrics 1998; 102(6): www.pediatrics.org/cgi/content/full/102/6/e6.5 screens.
74. Nakamura Y, Yanagawa H, Harada K, Kato H, Kawasaki T. Mortality among persons with a history of Kawasaki disease in Japan. Arch Pediatr Adolesc Med 2002; 156: 162-5.
75. Hirata S, Nakamura Y, Matsumoto K, Yanagawa H. Long-term consequences of Kawasaki disease among first-year junior high school students. Arch Pediatr Adolesc Med 2002; 156: 77-80.
76. Takechi N, Seki T, Ohkubo T, Ogawa S. Dobutamine stress surface mapping of myocardial ischemia in Kawasaki disease. Pediatrics International 2001; 43: 218-25.
77. Dajani A, Taubert K, Takahashi M, Bierman F, Freed M, Ferrieri P, et al. Guidelines for long-term management of patients with Kawasaki disease. Circulation 1994; 89: 916-22.
Correspondencia: Dr. Javier Prego Petit.
Hermanos Ruiz 3427. Montevideo, Uruguay.
E-mail: jotapre@adinet.com.uy


{kind=link}
{kind=link}