











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
Las amiloidosis son patologías infrecuentes causadas por proteínas anormalmente plegadas que se depositan en diversos órganos, fundamentalmente en el corazón y en el riñón. La más frecuente está causada por cadenas livianas libres de inmunoglobulinas producidas por plasmocitos aberrantes en la médula ósea. En las últimas décadas, se han incorporado herramientas de diagnóstico, tanto en la evaluación inmunoproteica como en la imagen cardíaca, que han mejorado la sospecha clínica y facilitado el seguimiento de compromiso de órgano. La espectrometría de masa se ha consolidado como el estándar para la tipificación de la proteína causante de la enfermedad. Además, han surgido nuevos tratamientos que modifican el curso de la enfermedad, interrumpiendo en diversas etapas el fenómeno de amiloidogénesis.
Nos enfocaremos en el presente artículo en el estudio diagnóstico desde el punto de vista hematológico.
Revisión del tema
Las amiloidosis son un grupo heterogéneo de patologías causadas por agregación y depósito de proteínas anómalas autólogas en distintos tejidos, sea de forma localizada o sistémica, adquirida o hereditaria1. Existen más de 30 proteínas patogénicas, y todas se caracterizan por el depósito de fibrillas con plegamiento anómalo, de entre 7 nm y 13 nm de diámetro en la microscopía electrónica. Además, se identifican componentes no fibrilares (glucosaminoglicanos) y amiloide sérico P en el depósito2. La estructura del amiloide otorga una propiedad característica en la tinción con Rojo Congo, que provoca una birrefringencia verde manzana cuando se observa con la luz polarizada. De todas las proteínas amiloidogénicas, las más frecuentes son las cadenas livianas de inmunoglobulinas (AL), la transtiretina (ATTR) en su forma nativa o mutada, y el amiloide sérico A (AA), vinculado a enfermedades crónicas inflamatorias o infecciosas1.
Las manifestaciones clínicas dependen del tipo de fibrilla depositada y el grado de compromiso tisular. Los dos subtipos más frecuentes (AL y ATTR) suelen generar compromiso cardíaco3.
La sospecha clínica suele surgir de la presencia de insuficiencia cardíaca (IC) con fracción de eyección (FEVI) preservada, hipertrofia ventricular izquierda (HVI) no explicada, y/o evidencia de compromiso de otros órganos, como proteinuria, insuficiencia renal, neuropatía periférica, trastornos digestivos, sangrado y/o disautonomía como síntomas frecuentes2,5.
El diagnóstico tardío se asocia con inferior sobrevida y calidad de vida6, por lo cual aumentar el índice de sospecha para detectarla en fase precoz es crucial.
Fisiopatología
Los mecanismos por los cuales las proteínas autólogas normales sufren un cambio conformacional hacia la forma patogénica son variados, involucrando aumento de síntesis por inflamación crónica o por discrasias de células plasmáticas, mutaciones de proteínas y envejecimiento, así como la disfunción de los sistemas de control del plegamiento proteico. El daño de órgano se genera por la precipitación y el depósito de las proteínas anómalas que provoca una alteración mecánica tisular, así como por citotoxicidad directa de las proteínas circulantes, causantes de inflamación, estrés oxidativo y activación de la apoptosis. La amiloidosis AL se debe a la producción de una proteína monoclonal inestable producida por plasmocitos aberrantes de la médula ósea. El compromiso cardíaco es frecuente, se observa en más de 50% de los casos; es el principal determinante de morbimortalidad de la enfermedad, con una sobrevida sin tratamiento inferior a 6 meses para AL y 3-5 años en ATTR7.
Manifestaciones clínicas
La presentación clínica suele ser inespecífica, lo cual determina un retraso diagnóstico superior a un año desde el inicio de los síntomas en el 40% de los pacientes8.
Los pacientes referidos a hematología son principalmente aquellos con sospecha de amiloidosis AL, usualmente derivados por presentar un proteinograma patológico en el estudio de manifestaciones no hematológicas o durante el seguimiento de una gammapatía monoclonal.
La amiloidosis AL representa el 71% de las amiloidosis, con una incidencia aproximada de 10 casos por millón de habitantes por año9. Es un trastorno de células plasmáticas clonales, escasamente proliferativas, productoras de fragmentos de cadenas livianas de inmunoglobulinas (FLC) inestables, habitualmente lambda (75%). Esas FLC generan toxicidad directa e infiltración de tejidos ocasionando su falla, con compromiso multiorgánico en la mayoría (69%) de los pacientes10.
La infiltración plasmocitaria medular suele ser baja (mediana 6-7%), y puede presentarse asociada a mieloma múltiple o a otras neoplasias de linfocitos B.
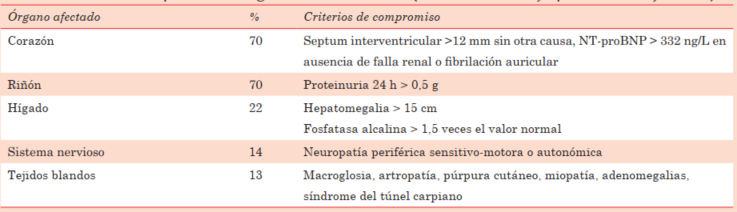
El compromiso cardíaco se presenta típicamente como una cardiomiopatía restrictiva, con IC sintomática (20% al debut), cardiomegalia y arritmias. Astenia, adinamia, fatiga e intolerancia al ejercicio son síntomas inespecíficos reportados con frecuencia. La hipotensión o intolerancia a antihipertensivos habituales son hechos frecuentes y de pobre pronóstico.
La afección renal está presente en casi 70% de los pacientes, y suele manifestarse por albuminuria o síndrome nefrótico, y en 45% con insuficiencia renal10.
El compromiso neurológico puede ocasionar una neuropatía sensitiva axonal, con afectación usualmente simétrica, distal de miembros, ocasionalmente dolorosa. La neuropatía autonómica se manifiesta por hipotensión ortostática, impotencia sexual y trastornos gastrointestinales (constipación o diarrea)10.
Puede ocurrir diarrea o sangrado por compromiso digestivo. La infiltración hepática se expresa por hepatomegalia y aumento de la fosfatasa alcalina. Otros signos clásicos son macroglosia, pseudohipertrofia muscular, aumento de partes blandas submandibulares, síndrome del túnel carpiano, púrpura facial y sangrados por deficiencia de factor X.
Los pacientes que al diagnóstico ya presentan compromiso significativo de órgano tienen inferior sobrevida y calidad de vida. En pacientes con gammapatía monoclonal de significado incierto (MGUS) con ratio FLC elevado, así como en pacientes con hipertrofia ventricular de causa no definida, la búsqueda de amiloidosis es necesaria para lograr un diagnóstico precoz, para lo que se recomienda el estudio sistemático con albuminuria, fosfatasa alcalina y NT-proBNP7.
Diagnósticos diferenciales
Amiloidosis por transtiretina: la transtiretina es una proteína de síntesis hepática, transportadora de tiroxina y vitamina A. Por envejecimiento o por mutaciones autosómicas dominantes esta proteína se disocia en productos intermedios que se agregan y depositan en diversos órganos, en particular en el corazón11.
Amiloidosis por transtiretina nativa (ATTRwt): se presenta como IC por cardiopatía hipertrófica, infiltrativa, con FEVI conservada hasta etapas muy avanzadas de la enfermedad. Es frecuente la disfunción autonómica. Esta entidad predomina en hombres, mayores de 60 años, afrodescendientes y es frecuente que asocien estenosis del canal lumbar, estenosis aórtica de bajo gradiente y bajo flujo, y rotura espontánea tendinosa12. Se estima que 10-15% de los hombres añosos con IC tienen una ATTRwt12. El centellograma miocárdico tiene una sensibilidad y valor predictivo positivo de 98% para diagnóstico de ATTRwt en pacientes sin componente monoclonal.
Amiloidosis por transtiretina hereditaria (ATTRh): existen más de 120 mutaciones patogénicas del gen de la transtiretina, con distribución geográfica marcada. La mutación Val30Met es la más frecuente en ciertas regiones (Suecia, Japón), y se presenta habitualmente como polineuropatía, si bien puede generar compromiso cardíaco. La variante ATTRh que más afecta el corazón es la Val122Ile, observada en 3-4% de la población afrodescendiente en Estados Unidos13. El diagnóstico se realiza por la secuenciación del gen ATTR o bien la búsqueda directa de la mutación, si se conocía previamente el antecedente familiar.
Dada la edad de presentación, las ATTR pueden asociar gammapatía monoclonal; la más frecuente es la gammapatía monoclonal de significado incierto. Más de 20% de los pacientes con ATTRwt presentan pico monoclonal en el proteinograma1. Para evitar un diagnóstico y tratamiento inadecuados la tipificación de la proteína causante es esencial.
Amiloidosis reactiva (AA): el compromiso cardíaco en amiloidosis AA puede observarse en fases muy avanzadas. Son pacientes con antecedente de una enfermedad inflamatoria con pobre control (por ejemplo, artritis reumatoidea, espondiloartropatías y enfermedades intestinales) y compromiso renal severo al momento en que el compromiso cardíaco se manifiesta14. Las patologías inflamatorias crónicas también pueden cursar con un componente monoclonal en el proteinograma, lo que genera dudas diagnósticas.
Valoración paraclínica
Ante la sospecha de amiloidosis, desde el punto de vista hematológico, los pasos para la evaluación son:
1. Diagnóstico y tipificación del amiloide.
2. Evaluación de compromiso de órganos.
3. Evaluación pronóstica.
1. Diagnóstico y tipificación del amiloide: el diagnóstico de amiloidosis exige su identificación en biopsia tisular. En AL, el aspirado de grasa subcutánea tiene una sensibilidad de 84-88%, la biopsia rectal 75-85%, la médula ósea 50% y las glándulas salivales menores 58%. La sensibilidad diagnóstica es muy inferior para las amiloidosis no AL. En un estudio de 216 pacientes con amiloidosis sistémica, la sensibilidad del aspirado de grasa subcutánea fue 84% para AL, 45% en ATTRh y 15% en ATTRwt15. Este procedimiento inocuo, de bajo costo y de realización ambulatoria, es el de elección para iniciar el estudio.
La biopsia del órgano afectado es la de mejor sensibilidad diagnóstica (> 90% renal, hepática y cardíaca); por su invasividad, se reserva para casos dudosos. En ocasiones, el paciente es derivado por el informe de biopsia de órgano, lo cual hace innecesarias las anteriores para el diagnóstico.
La muestra debe ser evaluada por patólogo entrenado. En la tinción con hematoxilina-eosina el amiloide se identifica como un depósito extracelular, amorfo, eosinófilo con birrefringencia verde manzana característica en la tinción con Rojo Congo, cuando se observa en el microscopio de luz polarizada.
Identificada la presencia de amiloide, se debe proceder a tipificar la proteína causante7. Las técnicas utilizadas son: inmunofluorescencia, inmunohistoquímica, inmunomicroscopía electrónica y espectrometría de masa.
Inmunofluorescencia: tiene un alto valor predictivo positivo (100%), pero la necesidad de evaluación en tejido en fresco y su baja especificidad la hacen poco prácticas.
Inmunohistoquímica: tiene alta sensibilidad para diagnóstico de AA y ATTR, pero no para AL, debido a que los cambios conformacionales de las proteínas la hacen menos reactivas a los anticuerpos utilizados16.
Inmunomicroscopía electrónica: tiene elevada sensibilidad, con la desventaja de ser una técnica laboriosa, artesanal, y lenta, disponible únicamente en centros de referencia17.
Espectrometría de masa: es el estudio ideal, identifica la proteína involucrada con alta sensibilidad (100%) y especificidad (98%). Tiene como desventajas su elevado costo, la baja disponibilidad y el requisito de realizar la microdisección láser de la zona infiltrada en la biopsia18. Por el momento, esta técnica no se encuentra disponible con este fin en Uruguay.
Estudios inmunoproteicos y de identificación de la clona plasmocitaria para el diagnóstico de amiloidosis AL:
Proteinograma electroforético en suero: permite evidenciar y cuantificar el componente proteico monoclonal, que se visualiza como pico o banda estrecha en el trazado.
Inmunofijación en suero: permite tipificar el componente monoclonal (IgG, IgA o IgM; kappa o lambda).
Cadenas livianas libres en suero: la inmunoglobulina monoclonal secretada por los plasmocitos puede ser una proteína intacta (inmunoglobulina entera) o solamente cadenas livianas (monómeros o dímeros de κ o λ). Habitualmente el componente monoclonal identificado en el proteinograma de las amiloidosis es pequeño, < 20 g/l en 70% de los pacientes, y en 20% no evidente. Las cadenas livianas libres son indispensables para el diagnóstico, tienen valor pronóstico y, por su corta vida media, son el parámetro requerido para controlar la respuesta al tratamiento.
La combinación de proteinograma electroforético e inmunofijación en suero y orina, y cadenas livianas libres en suero logran una sensibilidad de 100% para identificar una proteína monoclonal en pacientes con amiloidosis AL.
Cuantificación de las inmunoglobulinas (IgG, IgA, IgM): permite detectar la presencia de inmunoparesis (descenso de 1 o más), asociado a mayor riesgo infeccioso.
Proteinuria de 24 h con proteinograma e inmunofijación: permite detectar, cuantificar y tipificar las proteínas involucradas, con valor pronóstico para el tiempo a la diálisis.
Detección de la clona plasmocitaria: se realiza mediante aspirado (mielograma) y biopsia de médula ósea, para evaluar el porcentaje de plasmocitos (> 10% es un parámetro adverso y condiciona el tratamiento), inmunofenotipo para confirmar la clonalidad (kappa o lambda), citogenético convencional y FISH para detección de aberrancias citogenéticas, de las cuales la más frecuente es la t(11;14), presente en 46% de las AL, y las de mal pronóstico la t(14;16), t(4;14) y del17p19.
La combinación de biopsia de médula ósea y biopsia de grasa subcutánea permite detectar el 87% de las amiloidosis.
En la (tabla 1) se muestran los criterios diagnósticos y en la (figura 1) un algoritmo de estudio sugerido ante sospecha de amiloidosis AL.

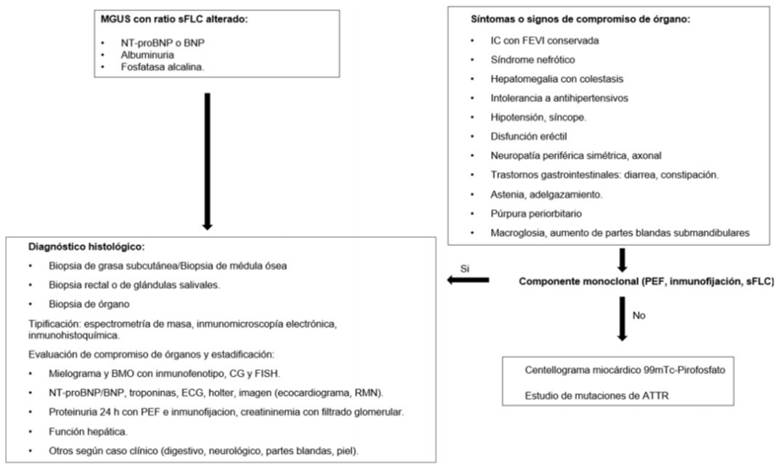
2. Evaluación de compromiso de órganos: incluye la valoración cardíaca, renal, neurológica, hepática, de partes blandas y digestiva, lo que será abordado en este especial (tabla 2).
Tabla 2: Criterios de compromiso de órgano en amiloidosis AL (XII International Symposium on Amyloidosis)
3. Evaluación pronóstica: la elección del tratamiento y el pronóstico vital dependen fundamentalmente de la severidad del compromiso cardíaco. En AL, la combinación de diferencial de FLC (involucradas - no involucradas) > 18 mg/dl, NT-proBNP > 1800 pg/ml y troponina T > 0,025 ng/ml permite diferenciar 4 grupos de riesgo en amiloidosis AL con sobrevida a 5 años entre 14% (3 factores presentes) y 59% (0 factor presente). El NT-proBNP > 8500 pg/ml se asocia con corta sobrevida, inferior a 6 meses, requerimiento de ajustes de dosis de tratamiento específico y contraindicación para trasplante de progenitores hematopoyéticos20,21.
El filtrado glomerular < 50 ml/min y la proteinuria > 5 g se correlacionan con la probabilidad de diálisis a 2 años (0%, 7% y 60% a 2 años), aunque sin impacto en la sobrevida global22.
El manejo terapéutico de la amiloidosis AL incluye tratamientos dirigidos a detener y/o revertir el proceso amiloidogénico, y tratamiento de soporte de los síntomas de compromiso sistémico, lo cual será abordado en este especial.
Conclusiones
Las amiloidosis son un grupo heterogéneo de enfermedades provocadas por el depósito de proteínas mal plegadas. La tipificación del amiloide es esencial para definir el tratamiento y el pronóstico.
La evaluación inmunoproteica y de médula ósea, así como los biomarcadores cardíacos, son esenciales para el adecuado manejo terapéutico.
Se debe estudiar amiloidosis en toda gammapatía monoclonal de significado incierto con ratio FLC elevado que asocie albuminuria, fosfatasa alcalina elevada o aumento del NT-proBNP, así como en pacientes con IC con FEVI conservada.

