











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Sra. Editora:
Introducción
El nacimiento anómalo de la arteria coronaria izquierda (ACI) desde la arteria pulmonar (AP), síndrome de ALCAPA (acrónimo de Anomalous origin of the Left Coronary Artery from the Pulmonary Artery), es una rara entidad que se presenta con una incidencia de 1 cada 300.000 nacimientos y corresponde a 0,25%-0,5% del total de las cardiopatías congénitas1,2.
Su importancia radica en la elevada mortalidad, más de 90% en los primeros días o semanas de vida3).
En el adulto la causa de muerte está vinculada a la insuficiencia coronaria (con sus diferentes formas de presentación) provocada por el fenómeno denominado “robo coronario”.
El diagnóstico se realiza por estudios de imagen y su tratamiento es la cirugía de la que están descritas varias técnicas.
Presentamos el primer reporte de un caso en Uruguay del síndrome de ALCAPA en el adulto. Se trata de una paciente en la que se sospechó el diagnóstico en el ecocardiograma transtorácico (ETT), se confirmó en la coronariografía (CACG) y se realizó cirugía exitosa con técnica de Takeuchi.
Caso clínico
Paciente de sexo femenino, 32 años, con antecedentes personales de hipertensión arterial. Consultó por historia de ángor de esfuerzo clase II. Al examen físico se destaca la presencia de soplo sistólico 2/6 en ápex con irradiación a axila. Se realizó prueba ergométrica graduada (PEG), que fue anormal por descenso de 1 mm del segmento ST al máximo esfuerzo en DI-AVL y de V4 a V6. Luego se realizó un ETT que informó: ventrículo izquierdo levemente dilatado, contractilidad sectorial normal, fracción de eyección de 56%; strain longitudinal global de -19, disminuido en los sectores basales de cara posterolateral y anterior, con contracción postsistólica de 33%; insuficiencia mitral (IM) leve. Severa dilatación de la arteria coronaria derecha (ACD), diámetro de 9 mm. A nivel del septum interventricular se observa flujo continuo, diastólico, que se dirige desde el septum posterior al anterior con velocidades elevadas (164 cm/s), y se interpreta debido a la presencia de arterias colaterales septales.
Con diagnóstico presuntivo de ALCAPA se realizó CACG que confirmó el nacimiento anómalo de la ACI desde la AP (figura 1).
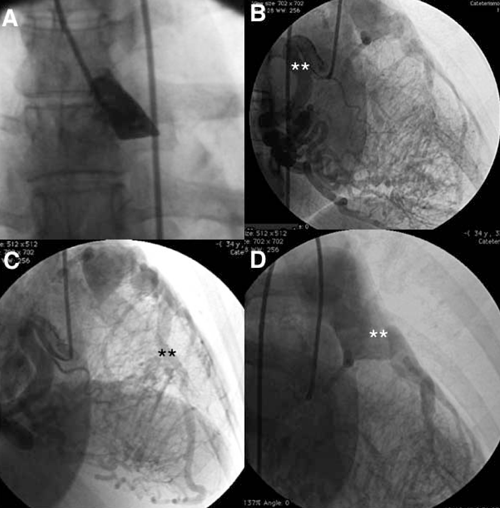
Figura 1: Coronariografía. A) Ausencia de la arteria coronaria izquierda en el seno de Valsalva izquierdo. B) Arteria coronaria derecha dilatada, flexuosa (**), con importante red de colaterales septales. C) Arteria descendente anterior ectásica (**), que recibe flujo desde la arteria coronaria derecha. D) Nacimiento de la arteria coronaria izquierda desde la arteria pulmonar (**).
La paciente fue intervenida quirúrgicamente mediante técnica de Takeuchi: se realizó abordaje por esternotomía, se resecó parte del pericardio y se preparó en gluteraldehído para su posterior utilización. A la inspección se destacaba el gran desarrollo de la ACD (figura 2A).
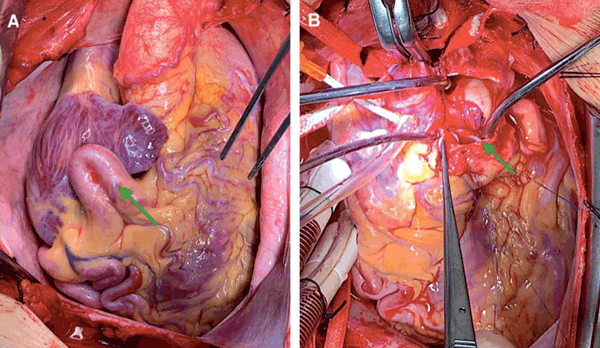
Figura 2: Imágenes del intraoperatorio. A) Se observa el gran desarrollo de la arteria coronaria derecha (flecha). B) Restauración del flujo coronario normal.
Para entrar en circulación extracorpórea se realizó doble canulación venosa (cavas) y arterial en aorta ascendente y se descendió la temperatura hasta alcanzar una hipotermia moderada. Posteriormente se realizó clampeo aórtico seguido de cardioplejia que fue administrada por la raíz aórtica, por la AP y por el ostium de la ACI de forma selectiva.
Primero se realizó la apertura anterior de la AP en forma de flap. Se identificó el ostium coronario en el seno izquierdo. Luego se hicieron dos orificios de 5 mm enfrentados, uno en la aorta y otro en la AP, y se suturó un borde con el otro, creando así una ventana aortopulmonar. Para la tunelización del flujo desde dicha ventana hasta el ostium izquierdo se suturó el flap a la pared posterior de la arteria pulmonar (figura 2B). Finalmente, se cerró el defecto de la cara anterior de la AP con el parche de pericardio autólogo, evitando de esta forma provocar una estenosis supravalvular pulmonar. Luego del declampeo aórtico, retomó actividad en ritmo sinusal y sin alteraciones en el ECG.
Discusión
Las anomalías coronarias se presentan con una incidencia de 1,3%, según diferentes series4. La mayoría cursa de forma asintomática; sin embargo, un porcentaje bajo se asocia a síntomas y complicaciones graves como angina, disnea, síncope, insuficiencia cardíaca y muerte súbita3. El síndrome de ALCAPA es una anomalía coronaria infrecuente con incidencia de 1 cada 300.000 nacimientos1. Su relevancia radica en la elevada mortalidad, que alcanza el 90% durante el primer año de vida, y en el riesgo de muerte súbita cardíaca (MSC) en el adulto, que es de 80%-90%, lo que hace su diagnóstico a esta edad excepcional3-5.
La mala evolución del síndrome de ALCAPA está asociada a la severa isquemia provocada por el fenómeno de “robo coronario”. Previo al nacimiento, las presiones en la AP son elevadas, por lo que el flujo en la ACI es anterógrado. Durante los primeros meses de vida la presión en la AP se reduce y el flujo sanguíneo se hace retrógrado, ocasionando una mala perfusión a nivel del ventrículo izquierdo con consecuencias graves, tales como infarto de miocardio, disfunción ventricular izquierda, isquemia silente, IM, arritmias malignas y MSC. En base a las formas de presentación y a la evolución, se describen dos patrones que están relacionados con el desarrollo de circulación colateral desde la ACD a la ACI. El denominado “patrón infantil” se caracteriza por ausencia o inadecuada circulación colateral, presencia de síntomas graves y mortalidad de 90% durante el primer año de vida. Por otro lado, en el denominado “patrón del adulto”, se observa un gran desarrollo de circulación colateral, suficiente para sobrevivir hasta la etapa adulta de forma asintomática; sin embargo, la mayoría de las veces los síntomas aparecen en la evolución y pueden presentarse complicaciones como las ya mencionadas3).
En un estudio en el que se describen las formas de presentación en el adulto se observó que el 18% de los pacientes presentaron arritmias malignas con riesgo de MSC, mientras que el 68% se presentó con síntomas como angina, disnea y palpitaciones, predominantemente. En relación con la paraclínica se observó que los estudios funcionales objetivaban isquemia en 87% de los casos y en 46% de éstos el ETT sugirió el diagnóstico6. En este caso la angina de esfuerzo fue la forma de presentación de la isquemia, que se demostró durante la realización del estudio funcional (PEG). A su vez, durante la realización del ETT la presencia de strain longitudinal disminuido en los sectores basales de la cara posterolateral y anterior contribuyó al diagnóstico. Adicionalmente se observó IM que se planteó, por el contexto, de etiología isquémica (provocada por la disfunción del músculo papilar). Los hallazgos ecocardiográficos fundamentales por los que se sospechó la enfermedad fueron: ACD con gran dilatación, presencia de circulación colateral objetivada por flujo que se dirigía desde el sector posterior del septum al anterior y flujo reverso en el territorio de la ADA.
El diagnóstico se confirma observando el origen anómalo de la ACI en la AP. Los estudios de imagen utilizados con este fin son la CACG, la angiotomografía computarizada (TC) y la resonancia nuclear magnética (RNM), cada uno con ventajas y desventajas. La CACG tiene como desventaja el ser invasiva, utilizar radiación ionizante y contraste, exponiendo al paciente a potenciales complicaciones. La TC tiene la ventaja de no ser invasiva y presentar una mayor disponibilidad. La RNM no es invasiva, no usa contrastes ni radiación y permite evaluar flujos, grado de IM, viabilidad del miocardio y fibrosis. En el caso que describimos se realizó CACG que mostró ausencia del origen de la ACI en el seno coronario izquierdo, opacificación retrógrada de la ACI, nacimiento de la ACI en la AP, importante desarrollo de colaterales de la ACD a la ACI y severa dilatación de la ACD, siendo elementos característicos7.
El tratamiento del síndrome de ALCAPA es quirúrgico. Se han descrito varias técnicas, siendo las más aceptadas por sus mejores resultados: el reimplante de la ACI en la aorta con la restauración de una circulación coronaria dependiente de un circuito de dos arterias8 y la ligadura del tronco coronario izquierdo con bypass de la arteria mamaria interna izquierda a la ADA. Debido a sus buenos resultados la técnica del reimplante de la ACI en la aorta es la de elección en la infancia, mientras que en el adulto se prefiere la ligadura de la ACI con la realización de bypass arterial o venoso9,10.
En este caso se realizó con éxito el reimplante del tronco coronario izquierdo en la aorta con técnica de Takeuchi.
Conclusiones
Las anomalías coronarias, que significan un verdadero riesgo para los pacientes, son infrecuentes. Dado que se pueden presentar en pacientes jóvenes con baja probabilidad de enfermedad coronaria, con síntomas y signos sugestivos de isquemia, se debe mantener un elevado nivel de sospecha.

