











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo

 Permalink
Permalink
Introducción
El hepatocarcinoma fibrolamelar (HCFL) es una variante rara del hepatocarcinoma, descrito por Edmondson en 19561. La variedad fibrolamelar se caracteriza por la presencia de bandas de colágeno fibrosas gruesas que rodean las células tumorales.
Su incidencia varía entre 1% y 5% en el porcentaje de todos los hepatocarcinomas. Afecta principalmente a pacientes jóvenes con hígado sano, entre los 10 y los 30 años, no habiendo diferencia entre ambos sexos1.
En el 50% de los casos el diagnóstico se realiza en etapas avanzadas de la enfermedad. En referencia a la patogénesis, en 2014, Honeyman y colaboradores estudiaron muestras de tumores de HCFL de 11 pacientes que expresaban una nueva transcripción de fusión, DNAJB1-PRKACA. El estudio de la secuenciación del genoma mostró que la fusión resultó de una dilección de aproximadamente 400 kilobases (kb) en el cromosoma 19. La transcripción de la fusión codifica una proteína quimérica que acopla un segmento de la proteína de choque térmico, DNAJB1, con el dominio catalítico de la proteína quinasa A (PKA) y muestra una retención total de la actividad PKA. En particular, la fusión DNAJB1- PRKACA no se detectó en ninguna de las muestras de tejido hepático no tumorales emparejadas2.
Múltiples estudios independientes confirmaron los hallazgos de Honeyman mediante la demostración de la presencia del gen de fusión en las HCF3,4. La falta aparente de una mutación de segundo éxito en el genoma de los HCFL respalda el papel de la proteína de fusión DNAJB1-PRACA como un impulsor principal de este tumor y lo señala como un objetivo diagnóstico y una posible diana terapéutica5.
En consecuencia, los HCFL mostraron una actividad PKA estimulada por monofosfato de adenosina cíclica (cAMP), en comparación con el tejido hepático normal, lo que sugiere que la señalización de PKA aberrante contribuye a la génesis tumoral hepática5.
La cirugía es el pilar actual del tratamiento y sigue siendo la única opción terapéutica potencialmente curativa. Las recurrencias son comunes, por lo que se continúan estudiando terapias alternativas.
Caso clínico
Sexo masculino, 15 años de edad. Procedente de Montevideo. Sin antecedentes personales patológicos.
Consulta por tumoración abdominal de tres meses de evolución, agregando, en los últimos 15 días antes de la consulta, dolor localizado en epigastrio, náuseas y vómitos.
Marcado adelgazamiento en los últimos meses.
De la paraclínica se destacaba: hemograma sin alteraciones. Funcional y enzimograma hepático alterado a expensas de una fosfatasa alcalina de 216 U/L, transaminitis leve con aspartato aminotransferasa (AST) de 80 U/L y alanino aminotransferasa (ALT) de 71 U/L, con una gamma glutamil transferasa (GGT) de 77 U/L.
Se solicitaron serologías para virus de hepatitis B (VHB) y virus de hepatitis C (VHC) negativas, con una alfafetoproteína (AFP) en rango de normalidad.
Examen físico
Se destaca a nivel abdominal una tumoración pétrea, topografiada en hemiabdomen superior de límites mal definidos, bordes irregulares, que ocupa todo el epigastrio, solidaria al hígado.
Ecografía de abdomen
Nódulo hepático heterogéneo anecoico, de paredes finas, de 87 por 69 mm en el lóbulo izquierdo compatible con nódulo sólido.
Tomografía computada
Hepatomegalia difusa de densidad heterogénea a expensas de múltiples lesiones sólidas, con realce en tiempo arterial; la de mayor tamaño ocupa la mayor parte del lóbulo izquierdo de 161 por 70 por 100 mm, de contornos irregulares (figura 1). Se observa en el segmento VI lesión nodular de 9 mm y en el segmento VII de 20 mm.
Adenomegalias y conglomerados a nivel intercavoaórticos de 43 por 42 mm, a la altura del hilio renal izquierdo, y lateoraórtico de 35 por 30 mm. Tórax: imagen nodular pleural derecha de 15 mm.
Resonancia magnética
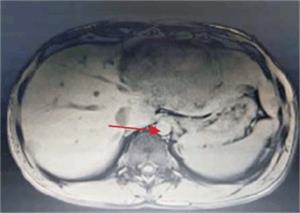
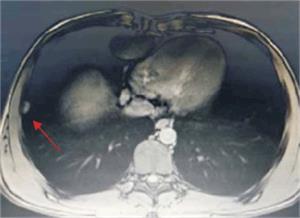
Tumoración que sustituye el lóbulo izquierdo con realce heterogéneo en la fase arterial que infiltra la vena suprahepática izquierda, contacta con la vena suprahepática media y la vena porta (figura 2) y (figura 3). Lesiones en los segmentos V y VIII. Extensas adenopatías en el hilio hepático. Nódulo peritoneal en fondo de saco de Douglas. Nódulo pulmonar derecho (figura 4).
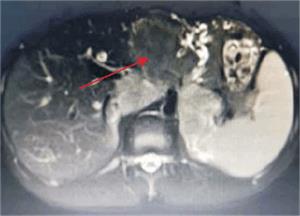
Figura 2: Tumor a nivel de lóbulo izquierdo con hipervascularización central. Realce de la vena porta.
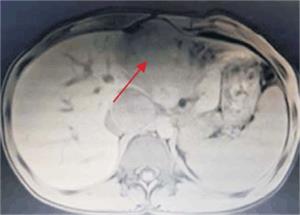
Figura 3: Se observa tumoración que protruye a nivel de epigastrio, palpable al examen físico abdominal.
Discusión
El HCFL es una variante poco frecuente del hepatocarcinoma, con una presentación clínica poco específica en etapas iniciales. La edad promedio de presentación es de 22 años, siendo una enfermedad estadio IV al momento del diagnóstico en el 50% de los casos. No presenta predominio por sexo1,6,7.
A diferencia del hepatocarcinoma, se presenta en pacientes jóvenes, sin hepatopatía previa1,6-8.
Característicamente las serologías para virus de hepatitis B y C son negativas, así como en la mayoría de los casos los marcadores tumorales no suelen estar elevados9-12.
Los estudios de imagen constituyen un pilar fundamental, siendo la confirmación diagnóstica definitiva anatomopatológica.
En la tomografía computada los hallazgos característicos son la presencia de lesiones grandes y heterogéneas, en su mayoría bien definidas y con bordes lobulados. La cicatriz central, que se observa en 65% a 70% de los casos, no es patognomónica; sin embargo, una cicatriz grande, con un diámetro mayor a 2 cm y la presencia de septos fibrosos, son más frecuentes en el HCFL respecto a otros hepatocarcinomas. La presencia de calcificaciones puede observarse en 40% a 68% de los casos. Puede observarse necrosis tumoral, pero la hemorragia intratumoral no es frecuente13,16.
En la resonancia magnética suele ser hipointenso en T1 e hiperintenso en T2. La cicatriz central fibrosa es típicamente hipointensa tanto en T1 como en T2.
Las metástasis linfáticas son más frecuentes y ocurren en 50% a 65% de los casos. Las linfoadenopatías se observan frecuentemente en el hilio hepático y en el ligamento hepatoduodenal, pero también pueden observarse en el retroperitoneo, la pelvis y el mediastino.
El compromiso nodal es un importante factor pronóstico13-16.
En cuanto a la anatomía patológica se trata de un tumor generalmente único, grande y duro, con bandas fibróticas que lo atraviesan; está constituido por células poligonales bien diferenciadas que se disponen en cordones separados por bandas de fibras de colágeno17.
Las metástasis a distancia fueron reportadas en 20% a 30% de los casos17-19.
La presencia de lesiones satélites dentro del hígado, la obstrucción biliar y la invasión vascular son factores asociados a peor pronóstico13,18,19.
Un porcentaje de los pacientes son candidatos a resección quirúrgica, con una tasa alta de recidiva a los diez años.
Las recurrencias son frecuentes y oscilan entre 33% y 100%, dependiendo de la duración, el seguimiento y el tipo de tratamiento quirúrgico instaurado19.
En términos generales, el HCFL presenta una menor respuesta al tratamiento oncoespecífico en contraposición al hepatocarcinoma convencional; sin embargo, la quimioterapia puede ser útil en casos avanzados20.
En cuanto al pronóstico, una revisión sistemática, que incluyó 35 casos, concluyó que tiene un mejor pronóstico que el hepatocarcinoma convencional21. Factores pronóstico asociados con HCFL incluyen edad, estadio de la enfermedad, multiplicidad, trombosis tumoral, invasión linfovascular, metástasis y si se realizó o no una resección quirúrgica completa22.
Otros autores, como Kaseb, encontraron que la raza blanca, el sexo femenino, el estadio tumoral temprano y la resección del tumor, incluida la metastasectomía, se asociaron positivamente con una sobrevida más larga. El sexo femenino fue el único factor predictivo positivo de una mayor supervivencia sin recidiva tumoral23.
Conclusión
El HCFL es una entidad poco frecuente. La mayoría de la evidencia científica proviene de reportes de casos clínicos o estudios con un número pequeño de pacientes. Se presenta en pacientes jóvenes, como en nuestro caso. A diferencia del hepatocarcinoma convencional, se diagnostica en pacientes sin factores de riesgo para cirrosis.
El diagnóstico suele realizarse en etapas avanzadas de la enfermedad. El pronóstico de estos pacientes es mejor en comparación con el hepatocarcinoma convencional.
El tratamiento quirúrgico es el más efectivo dada la poca respuesta a la terapia oncoespecífica. Los recientes hallazgos de una fusión intracromosómica recurrente gene-DNAJB1-PRKACA, que es responsable de la patogénesis de este tumor, propone nuevos desafíos a la investigación de posibles dianas terapéuticas.

