










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkRevista Médica del Uruguay
versão On-line ISSN 1688-0390
Rev. Méd. Urug. vol.28 no.3 Montevideo set. 2012
Estudios de bioequivalencia in vivo para demostrar la intercambiabilidad de medicamentos
Dres. Francisco Estévez*, Susana Parrillo†, Br. Q.F. Mónica Cedrés‡
Centro de Investigación en Farmacología Clínica-Bdbeq SA, Hospital Italiano. Montevideo, Uruguay
* Director. Centro de Investigación en Farmacología Clínica-Bdbeq SA, Hospital Italiano, Montevideo, Uruguay. Director del Master en Farmacología Clínica, Centro de Ciencias Biomédicas, Universidad de Montevideo. Ex Prof. Agdo. del Departamento de Farmacología y Terapéutica, Facultad de Medicina, Universidad de la República. Uruguay
† Jefe de la Unidad Clínica. Centro de Investigación en Farmacología Clínica-Bdbeq SA, Hospital Italiano, Montevideo, Uruguay. Ex Profesor Adj. Cátedra de Farmacología y Biofarmacia. Facultad de Química y Farmacia. Universidad de la República. Uruguay.
‡ Jefe de la Unidad Bioanalítica. Centro de Investigación en Farmacología Clínica-Bdbeq SA, Hospital Italiano, Montevideo, Uruguay.
Correspondencia: Dr. Francisco E. Estévez Carrizo. CIFC-Bdbeq SA, Hospital Italiano, primer piso. Br. Artigas 1632. Montevideo, Uruguay. Correo electrónico: francisco.estevez@bdbeq.com.uy
Conflicto de intereses: Los autores del presente artículo declaran que no existen conflictos de intereses.
Recibido: 8/8/12. Aceptado: 28/9/12
Resumen
Introducción: la exigencia de demostración de la intercambiabilidad de medicamentos genéricos en relación con los innovadores comenzó en el mundo desarrollado en la década de 1970. Objetivo: en el presente artículo se hace una breve reseña histórica señalando los medicamentos con los cuales y por primera vez se constataron problemas de bioequivalencia. Resultados: se señala que la implementación de la intercambiabilidad en América Latina comienza a principios de la primera década de este siglo, impulsado por un grupo de trabajo de la Organización Panamericana de la Salud (OPS), y que Uruguay comenzó a legislar en la materia en enero de 2007. Se definen los conceptos de bioequivalencia y de intercambiabilidad de medicamentos y se aclara cuáles son los medicamentos que deberían cumplir con esta exigencia en nuestro país. Se examinan los criterios para diseñar un listado de prioridades para exigir la bioequivalencia y se hace una revisión del marco regulatorio que nuestro país se ha dado desde el año 2007. Se hace referencia a diseños específicos de estos estudios cuyo objetivo es evaluar la interacción de las nuevas formulaciones "retard" con la comida o minimizar el efecto de la variabilidad intraindividual sobre la potencia estadística de los estudios. Conclusiones: se comentan algunos detalles mejorables de la normativa y el impacto de los estudios de bioequivalencia sobre los hábitos de prescripción, el mercado farmacéutico en Uruguay y la accesibilidad a medicamentos eficaces y seguros.
Palabras clave: EQUIVALENCIA TERAPEUTICA INTERCAMBIABILIDAD DE MEDICAMENTOS
Keywords: THERAPEUTIC, EQUIVALENCY INTERCHANGE OF DRUGS
Introducción
La exigencia de demostración de la intercambiabilidad de medicamentos genéricos en relación con los innovadores (originales) comenzó en el mundo desarrollado en la década de 1970. Digoxina(1), fenitoína(2,3), ciclosporina(4), warfarina(5), teofilina(6), fenotiazinas(6), las preparaciones de tiroides(6), los estrógenos conjugados(6), etcétera, son ejemplos históricos de medicamentos que han tenido "problemas de biodisponibilidad", con consecuencias de relevancia clínica: falta de efecto o toxicidad.
El objetivo del presente artículo es presentar una revisión de la evolución normativa y metodológica de la intercambiabilidad de medicamentos genéricos en el mundo, en la región y, particularmente, en nuestro país. Además, se establecen algunos puntos de comparación entre países de la región y con organizaciones normativas internacionales.
Breve reseña histórica
A comienzos de los años 70, algunas de las moléculas mencionadas más arriba, particularmente digoxina y fenitoína, alertaron sobre el problema y dieron lugar a la instauración de la exigencia de demostración de la bioequivalencia (BE) en países como Inglaterra, Canadá y Estados Unidos. Además, con el correr de los años se han ido sumando nuevas naciones y nuevos grupos terapéuticos.
A fines de la década de 1990 un grupo de trabajo de la OPS comenzó a promover el desarrollo de las actividades de BE en América Latina (Pan American Network for Drug Regulatory Harmonization [PANDRH], Working Group/Bioequivalence [WG/BE])(7). Los esfuerzos pioneros, ya en la primera década de este siglo, fueron de Argentina, Brasil y México. Los demás países de la región se encuentran en diferentes fases del desarrollo y aplicación efectiva de la normativa. La Organización Mundial de la Salud (OMS), en al año 2005, redactó unas guías para unificar los estándares de las organizaciones que llevan a cabo estudios de BE in vivo(8).
Definiciones
La biodisponibilidad (BD) se define como la fracción de la dosis administrada que efectivamente se absorbe y llega al medio interno (sangre sistémica) y la velocidad con que este proceso se verifica. El área bajo la curva concentración del fármaco versus tiempo (ABC) es proporcional a la fracción absorbida y la concentración máxima (Cmáx) refleja la velocidad de absorción. La BD absoluta es la comparación entre las ABC, por la vía oral (o cualquier otra vía que implique absorción) y la vía intravenosa (no hay absorción): ABCvo/ABCiv.
La BE es una forma de BD relativa en la cual se comparan dos formulaciones de un mismo fármaco (de distintos fabricantes) administrados por la misma vía. Se calcula de la siguiente manera: ABCT/ABCR (siendo T un medicamento de prueba y R un medicamento de referencia). Cuando este cociente ABCT/ABCR = 1 podemos decir que ambas formulaciones se absorben en la misma medida.
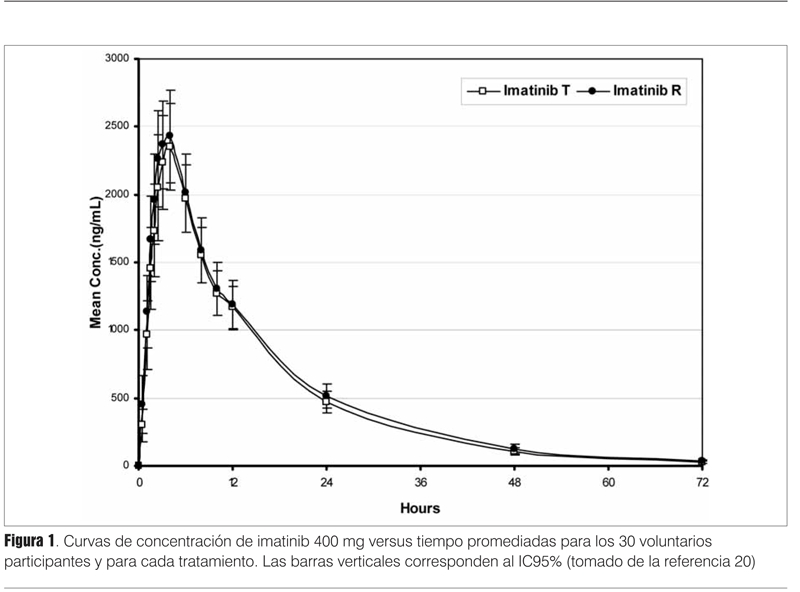
En la figura 1 se pueden apreciar las curvas concentración versus tiempo de 30 voluntarios sanos que tomaron dos formulaciones de un inhibidor de la tirosinfosfoquinasa (imatinib). Como se puede apreciar ambas curvas son prácticamente superponibles, por lo tanto ambas ABC promediadas son iguales. Sin embargo, esto no basta para concluir la BE. Como los estudios se hacen en una población de individuos, el otro factor que se debe tomar en cuenta es la variabilidad entre las formulaciones que se comparan.
Se debe hacer notar que los estudios de BE no son privativos de los medicamentos genéricos (en este artículo "genérico", "genérico de marca" y "similar" se usan como sinónimos). Los productos de investigación & desarrollo (I&D), comúnmente denominados "originales" o "innovadores", también deben demostrar la intercambiabilidad con estudios de BE durante el desarrollo clínico(9).
Medicamentos intercambiables
Un nuevo producto farmacéutico oral que tiene el mismo principio activo, a la misma dosis y en la misma forma farmacéutica (comprimidos, cápsulas, etcétera) que un producto de I&D, se denomina "equivalente farmacéutico". Cuando a dos "equivalentes farmacéuticos" se les realiza estudios que comparan sus biodisponibilidades en seres humanos y cumplen con ciertos criterios regulatorios de equivalencia se les denomina bioequivalentes.
El concepto de intercambiabilidad se basa en el siguiente presupuesto: si dos equivalentes farmacéuticos son bioequivalentes, las concentraciones del fármaco se equilibran en el organismo sincrónicamente, por lo tanto la evolución de las concentraciones en la biofase (compartimiento receptorial) va a ser prácticamente la misma para ambos productos. En este caso el efecto farmacológico es el mismo y, por ende, se puede inferir la equivalencia terapéutica y los productos serían intercambiables(10).
Los productos similares que se producen en nuestro país son "equivalentes farmacéuticos" de los respectivos innovadores. Sin embargo, en la mayoría, aún no se han implementado los estudios para demostrar la intercambiabilidad. La (tabla 1) muestra las características de los productos a los cuales hay que hacerles estudios de BE. Por otra parte, actualmente se comercializan en Uruguay productos farmacéuticos importados de otras regiones del mundo. Estos productos pueden tener estudios de BE hechos en el país de origen y, por lo tanto, ser intercambiables para esas poblaciones, sin embargo nuestra legislación exige que los estudios se realicen en nuestra población.

Aun cumpliendo con los requisitos farmacopeicos in vitro, no siempre se puede predecir el comportamiento terapéutico de equivalentes farmacéuticos de diferente procedencia. Tanto es así que cuando una compañía farmacéutica de I&D cambia el lugar de fabricación de uno de sus productos patentados (por ejemplo, de Suiza a Colombia), debe hacer estudios de BE para demostrar que, a pesar del cambio del lugar de fabricación, la eficacia terapéutica y la seguridad del producto siguen siendo las mismas (o sea, que el producto original es intercambiable consigo mismo)(11).
Criterios para la confección del listado de prioridades
El grupo de trabajo de la OPS recomendó adoptar dos criterios para seleccionar, caso a caso, los medicamentos para confeccionar una lista de prioridades. El primer criterio fue el de "riesgo sanitario" del medicamento involucrado, y el segundo criterio fue la "clasificación biofarmacéutica" (CBF), que ordena a los fármacos por su solubilidad en agua y permeabilidad a través del epitelio gastrointestinal.
A modo de ejemplo: es de suma importancia conocer si una nueva formulación de un genérico inmunosupresor (por ejemplo, tacrolimus) es intercambiable con el original. En caso de falla en la BE existe un alto riesgo sanitario, ya que se pone en juego la viabilidad del órgano trasplantado con el consecuente riesgo vital para el paciente. Por otra parte, el tacrolimus es un medicamento con baja solubilidad en agua (mala CBF). La ponderación conjunta de ambos criterios determina, en este caso, que el medicamento deba cumplir con la exigencia de BE sin posibilidad de bioexención (eximirse del estudio in vivo mediante un estudio in vitro).
Es interesante apuntar que los criterios aplicados por agencias regulatorias como la FDA (Estados Unidos)(12), la EMA (Unión Europea)(13,14) o ANVISA (Brasil), son diferentes (en realidad opuestos) a los recomendados por el grupo de trabajo en BE de la OPS-OMS para América Latina(7,8). En estos países de alta vigilancia sanitaria, en principio se exigen estudios de BE a todos los productos que sufren absorción para actuar a nivel sistémico. A solicitud del fabricante, la autoridad sanitaria puede eximir del estudio in vivo (bioexención) al medicamento cuyo principio activo demuestre alta solubilidad y/o permeabilidad en el tracto gastrointestinal y bajo riesgo sanitario.
A pesar de las diferencias de criterios, todas las agencias regulatorias que conforman la Conferencia Internacional de Armonización (CIARM o ICH) coinciden en que la BE es la herramienta idónea para demostrar la intercambiabilidad de los medicamentos genéricos que actúan a nivel sistémico, tienen un estrecho margen terapéutico y/o problemas de solubilidad/permeabilidad(12-14). Por lo tanto, es auspicioso que Uruguay haya legislado en la materia y esté dando los primeros pasos en la exigencia de estos estudios.
Marco regulatorio en Uruguay
Intercambiabilidad de medicamentos
En Uruguay, la intercambiabilidad de medicamentos ha sido reglamentada por el Ministerio de Salud Pública (MSP) desde enero de 2007 (decreto 12/007)(15). Este decreto aprueba las recomendaciones técnicas para la realización de estudios de BE y el cronograma operativo para la implementación de la exigencia de los estudios de BE.
El artículo 15° de dicho decreto establece que la equivalencia biofarmacéutica (BE) deberá demostrarse in vivo (con su correspondiente correlación in vitro) o, en algunos casos, in vitro mediante test de disolución. A esto último se le denomina bioexención. Los requisitos para determinar si los estudios se deben realizar in vivo o in vitro se establecen teniendo en cuenta el "riesgo sanitario" y la "clasificación biofarmacéutica" de cada fármaco.
Este decreto especifica cuáles deben ser los parámetros farmacocinéticos que se utilizan para demostrar la BE (ver tabla 2). Estos parámetros miden la cantidad del fármaco que se absorbió (ABC) y la velocidad con que se absorbe (Cmáx).

En el Anexo III se muestra un listado de los fármacos con prioridad a ser evaluados (ver tabla 3). Este listado fue publicado en enero de 2007 y se establece que será actualizado periódicamente por una comisión nombrada a tales efectos. Los medicamentos de liberación prolongada, comúnmente llamados retard, deben cumplir con el requisito de intercambiabilidad en todos los casos, esto es, independientemente de si su principio activo está o no en el listado del Anexo III. Por otra parte, la normativa uruguaya, siguiendo las pautas regulatorias internacionales(15,16), exigen dos estudios de BE a los productos LP, uno en ayunas y otro en estado posprandial inmediato, a efectos de descartar interacción entre la comida y la nueva formulación LP ("retard"). La normativa argentina exige un solo estudio en estado posprandial inmediato(17) para los productos de liberaci&oacut e;n prolongada.

El decreto también establece que la demostración de la BE se debe realizar en el país excepto que no se contara con la infraestructura adecuada, en cuyo caso el MSP autorizará la realización del estudio en otro estado parte del Mercosur.
Por último, cuando un producto (test) haya demostrado que es bioequivalente con el innovador (referencia) mediante estudios de BE realizados en un centro habilitado por el MSP, deberá llevar en el rótulo del envase la leyenda "medicamento intercambiable".
Aspectos éticos de la investigación clínica
El decreto 379/008, del 4 de agosto de 2008(18), comienza diciendo en el capítulo I -Finalidad, Términos y Definiciones-: "Esta ordenanza tiene por finalidad la protección integral de los seres humanos sujetos de una investigación, con especial consideración por su dignidad e integridad". El decreto establece que la libertad para llevar a cabo una investigación en seres humanos tiene como límite la libertad y los derechos esenciales que emanan de la persona, reconocidos en la Constitución de la República y en los tratados internacionales ratificados por la República y que se encuentren vigentes.
La normativa dispone que previo a toda investigación clínica -por ejemplo, los estudios de BE- se deberá contar con la aprobación del comité de ética institucional, el que deberá ser acompañado de sus respectivos protocolos de investigación y consentimiento informado.
En el Anexo se tratan diversos temas que tienen que ver con los aspectos éticos de la investigación que involucra a seres humanos, en especial el consentimiento libre e informado, el protocolo de investigación, la organización y las funciones de los comités de ética en investigación institucionales y las funciones y prerrogativas del Comité Nacional de Ética en Investigación (composición, atribuciones, etcétera) que depende de la Dirección Nacional de Salud del MSP.
Centros para el estudio de la bioequivalencia
El decreto 261/009, del 1º de junio del 2009(19), en su Anexo define: "Centro para Estudios de Biodisponibilidad y Bioequivalencia de Medicamentos" como la institución de investigación de carácter público o privado, habilitada por el MSP, en la que se realizan ensayos de BE in vivo (en seres humanos sanos o pacientes).
En el mismo Anexo se define que el director debe ser un médico o químico farmacéutico responsable por todas las actividades desarrolladas en el centro. Se define al investigador principal (IP) como "el médico o químico farmacéutico, especialmente calificado en el área, responsable del diseño de un protocolo de investigación específico, de la coordinación y la ejecución de la correspondiente investigación, así como por la integridad y derechos de los sujetos participantes de la misma", y se dice que el director será en todos los casos el IP.
El director/IP debe notificar al MSP de la realización de un estudio o solicitar autorización para la realización del mismo, de acuerdo a que el estudio requiera o no autorización previa. Los estudios con productos farmacéuticos cuyos principios activos son los fármacos listados en el Anexo III del decreto 12/007 (tabla 2), no requieren autorización previa, todos los demás sí la requieren.
El decreto define el ámbito de responsabilidades del investigador clínico (IC), del investigador del área bioanalítica, del investigador del área estadística, del responsable de la gestión de calidad y del promotor del estudio. Por otra parte, el Anexo del decreto describe las condiciones regulatorias de la organización y gestión de un centro, del protocolo de estudio, de la fase clínica, de la fase bioanalítica, de la fase farmacocinética y cálculos estadísticos y del informe final del estudio.
Lista de referencias para los estudios de bioequivalencia
La referencia es el medicamento que se utiliza como comparador del equivalente farmacéutico del cual se quiere demostrar la BE. Normalmente, las referencias son los respectivos medicamentos de I&D que han demostrado la eficacia y la seguridad en estudios clínicos aleatorizados y multicéntricos, y que han obtenido la licencia de comercialización de una agencia regulatoria miembro de la ICH (International Conference on Harmonization).
El decreto del MSP del 13 de diciembre de 2010(20) dispone las "Referencias para la realización de estudios de biodisponibilidad y bioequivalencia de medicamentos". En este decreto se encuentra un listado de las referencias para los fármacos mencionados en el Anexo III del decreto 12/007. Además, se establecen los "Criterios para la definición de referencias para la realización de estudios de biodisponibilidad y bioequivalencia de medicamentos" para los fármacos que no están comprendidos en el listado.
Plazos y mecanismo para realizar estudios en el exterior
Existe otro decreto de marzo de 2011(21) que actualiza los plazos para el cumplimiento de lo estipulado en el decreto 12/007 (que en los hechos vencieron) y, además, establece el mecanismo para realizar estudios de BE en estados parte del Mercosur, en caso de que la capacidad instalada en el país fuera insuficiente.
Guías de bioequivalencia
Por último, el MSP está elaborando las "Guías para la realización de estudios de biodisponibilidad/bioequivalencia", las "Guías para la realización de estudios de equivalencia biofarmacéutica in vitro", y los "Criterios para medicamentos referencia nacional". Estas guías describen en forma más extensa algunas directivas establecidas en los decretos reglamentarios y, particularmente, confieren un mayor grado de elasticidad al espónsor y/o centro de estudios de BE para presentar alternativas metodológicas ad referéndum de la autoridad sanitaria.
Aspectos metodológicos de los estudios
El detalle del diseño, ejecución y los cálculos farmacocinéticos/estadísticos de estudios realizados en nuestro país se pueden ver en detalle en artículos publicados(22-25). En esta sección se van a plantear algunos problemas que surgen con la variabilidad farmacocinética en las poblaciones estudiadas y los recursos metodológicos que se aplican para controlarla.
La variabilidad entre individuos (CVb) y dentro de cada individuo (CVw) es un factor muy importante en los estudios de BE. La variabilidad se constata en el ABC (cantidad absorbida) y en el Cmáx (velocidad de absorción). En este último caso existen parámetros alternativos a la Cmáx que mejoran la precisión y exactitud de la medición de la velocidad de absorción, particularmente para medicamentos LP, por ejemplo, el índice Cmáx/ABC(26,27). Sin embargo, el perfeccionamiento del instrumento de medición no controla toda la variabilidad.
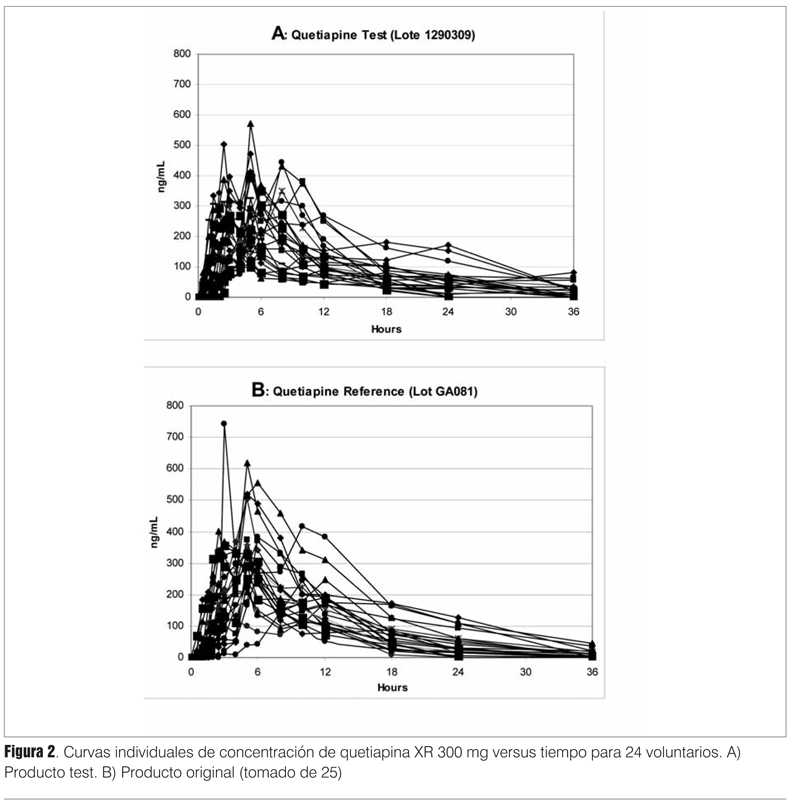
Las figuras 2A y 2B muestran las curvas individuales de un estudio de 24 voluntarios sanos graficadas por formulación (test y referencia). La quetiapina es un fármaco de variabilidad "moderada", su CVw varía en el entorno de 20%(24). Cuando un fármaco presenta un CVw mayor a 30%, se define como de "alta variabilidad"(10,11) y en ese caso se deben extremar las medidas para lograr una potencia estadística aceptable (>80%) con el menor número posible de sujetos de investigación.
Son múltiples los factores que determinan la variabilidad farmacocinética intraindividual, a saber: fisiológicos, patológicos, dietéticos, genéticos, etcétera. Salvo los factores genéticos cuya tipificación aún no se ha generalizado, los demás factores causantes de variabilidad farmacocinética se controlan en estudios de BE(28-30).
Los abordajes para controlar estos factores son fundamentalmente dos: A) las condiciones del estudio, y B) el diseño del estudio. El control de las condiciones del estudio (por ejemplo, factores que hacen variar el clearance sistémico del fármaco) minimiza el CVw de la población reclutada. Por otro lado, el diseño cruzado elimina el CVb poblacional del cálculo de la BE.
A estos dos pilares para controlar la variabilidad individual se le agrega un tercer pilar metodológico -la aleatorización de las secuencias de tratamientos- que compensa los posibles errores sistemáticos entre períodos de estudio.
Comentarios y discusión
Existen algunos puntos en la normativa vigente sobre la intercambiabilidad de genéricos que merecen comentarios, a saber: el director del centro debería ser siempre un médico, en lo posible, especializado en farmacología clínica. Este profesional es director del centro e IP de los estudios al mismo tiempo y, por lo tanto, debe responder (técnica, legal y deontológicamente) por el diseño, ejecución y reporte del estudio así como por la integridad física, psíquica, y el bienestar espiritual de los participantes. Es opinable que esta responsabilidad la pueda tomar un profesional que no está habilitado para ejercer la medicina ni tenga formación en investigación clínica.
Se debería revisar la lista de fármacos con prioridad a ser evaluados a efectos de actualizarla. Hay medicamentos que figuran en la lista que han quedado obsoletos y caído en desuso (por ejemplo, algunos antirretrovirales) y, por otro lado, faltan otros de amplio uso clínico y alto riesgo sanitario (anticoagulantes orales, inmunosupresores, antiepilépticos, inhibidores de la tirosinquinasa, heparinas de bajo peso molecular, antidiabéticos orales, antidepresivos, antihipertensivos, etcétera).
La BE está dando los primeros pasos en el país. A corto-mediano plazo, dependiendo de la voluntad política existente, se hará patente la gradual transformación del arsenal terapéutico nacional en la medida en que una porción cada vez mayor del mercado de los medicamentos similares ostente la demostración de la intercambiabilidad in vivo.
En este proceso habrá una transformación gradual del vademécum; muchas presentaciones farmacéuticas que hoy compiten por un lugar en el mercado se verán obligadas a dejar el lugar a productos con BE -intercambiabilidad- demostrada. La consecuencia final de esta nueva realidad será un mercado farmacéutico con amplia participación de los medicamentos similares intercambiables que competirán en condiciones más sanas y favorables con los productos innovadores. Sin embargo, lo más importante desde el punto de vista sanitario es que la población dispondrá de medicamentos seguros, eficaces y accesibles.
Además, en la medida que vaya desapareciendo del imaginario del colectivo médico la duda sobre la eficacia y la seguridad que hoy genera la prescripción de medicamentos sin demostración de intercambiabilidad, cambiará la actitud y los hábitos de los colegas. Este será el contexto sociocultural donde se podrán sentar las bases para la concreción de un uso más racional y eficiente del medicamento.
Por último, el establecimiento de un sistema regulatorio y fiscalizador de los estudios de BE y de los centros donde se llevan a cabo, y, por otro lado, el fomento y la promoción activa de los medicamentos genéricos por parte del Estado, constituirán una parte sustancial de la política nacional de medicamentos.
Abstract
Introduction: the demand to prove the interchangeability of generic drugs with innovative medicines originated in 1970, in the developed world.
Objective: the present article comprises a brief historical summary, pointing out the first drugs that caused bioequivalence problems.
Results: the study states the implementation of interchangeability in Latin America started the first decade of the current century, encouraged by a working team of the PAHO, and in January, 2007, Uruguay passed the first laws on this matter.
The concepts of drug bioequivalence and interchangeability are defined and the drugs that should observe this regulation are mentioned. The criteria for the design of a priorities list for the demand of bioequivalence are examined, and a review of the regulatory framework in our country since 2007 is also included in the present study.
Reference to specific designs of these studies is made, aiming to assess the interaction of the new retard formulations with food or to minimize the effects of intra-individual variability on the statistical value of the studies.
Conclusions: a few improvable details in terms of regulation and the impact of bioequivalence studies on prescription habits, the pharmaceutical market in Uruguay and accessibility to effective and safe drugs are commented upon.
Resumo
Introdução: a exigência da demonstração da intercambiabilidade de medicamentos genéricos em relação aos medicamentos de referência começou no mundo desenvolvido na década de 70.
Objetivo: neste artigo faz-se uma breve resenha histórica indicando os medicamentos com os quais se constataram problemas de bioequivalência pela primeira vez.
Resultados: a implementação da intercambiabilidade na América Latina começou nos primeiros anos deste século, impulsada por um grupo de trabalho da Organização Panamericana da Saúde (OPAS); o Uruguai começou a legislar sobre esse tema em janeiro de 2007.
Definem-se os conceitos de bioequivalência e de intercambiabilidade de medicamentos e se especifica quais são os medicamentos que deveriam cumprir com esta exigência no nosso país. Faz-se uma análise dos critérios utilizados para definir uma lista de prioridades para exigir a bioequivalência e uma revisão do quadro regulatório vigente no Uruguai desde 2007.
Faz-se referencia aos protocolos específicos dos estudos cujo objetivo é avaliar a interação das novas formulações "retard" com os alimentos ou minimizar o efeito da variabilidade intraindividual sobre a potencia estatística dos estudos.
Conclusões: discutem-se alguns detalhes da legislação que podem ser melhorados e o impacto dos estudos de bioequivalência sobre os hábitos de prescrição de medicamentos, o mercado farmacêutico no Uruguai e o acesso a medicamentos eficazes e seguros.
Bibliografía
1. Jounela AJ, Sothmann A. Bioavailability of Digoxin. Lancet 1973; 1(7796):202-3.
2. Albert KS, Sakmar E, Hallmark MR, Weidler DJ, Wagner JG. Bioavailability of diphenylhydantoin. Clin Pharmacol Ther 1974; 16(4):727-35.
3. Soryal I, Richens A. Bioavailability and dissolution of proprietary and generic formulations of Phenytoin. J Neurol Neurosurg Psychiatry 1992; 55(8):688-91.
4. Bennett WM, DeMattos A, Norman DJ, Meyer MM, Olyaei A. Which Cyclosporin formulation? Lancet 1996; 348(9021):205.
5. Vercaigne LM, Zhanel GG. Clinical significance of bioequivalence and interchangeability of narrow therapeutics range drugs: focus on warfarins. J Pharm Pharm Sci 1998; 1(3):92-4.
6. Ross MB. Status of generic substitution: problematic drug classes reviewed. Hosp Formul 1989; 24(8):441-4, 447-9.
7. Organización Panamericana de la Salud. Criterios científicos para los ensayos de bioequivalencia (in vivo e in vitro), las bioexenciones y las estrategias para su implementación: documento borrador. En: IV Conferencia Paneamericana para la Armonización de la Reglamentación Farmacéutica. Republica Dominicana 2-4 de Marzo , 2005. p.6. Disponible en: http://www.paho.org/spanish/ad/ths/ev/bedocumentocientificoborradorespanol.pdf [Consulta: 11 de jul de 2012].
8. World Health Organization. Additional guidance for organizations performing in vivo bioequivalence studies. In: Fortieth report of the WHO Expert Committee on Specifications for Pharmaceutical Preparations: Annex 9. Technical Report Series, No. 937. Geneva: WHO, 2006.
9. European Medicines Agency. ICH Topic E 6 (R1):guideline for good clinical practice. Disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdf [Consulta: 6 de jul de 2012].
10. Hauschke D, Steinijans V, Pigeot I. Bioquivalence studies in drug development: methods and applications. West Sussex, England: Wiley, 2007.
11. Schulz HU, Steinijans VW. Striving for standards in bioquivalence Assessment: a review. Int J Clin Pharmacol Ther Toxicol 1992; 30 (Suppl. 1):S1-6.
12. United States. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products. General considerations. Washington, DC: CDER, 2003. Disponible en: http://www.fda.gov/downloads/ Drugs/.../Guidances/ucm070124.pdf. [Consulta: 11 de jul de 2012].
13. European Medicines Agency. Note for guidance on the investigation of bioavailability and bioequivalence. London: EMA, 2001. Disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003008.pdf [Consulta: 11 de jul de 2012].
14. Estévez Carrizo FE. Estudios de bioequivalencia: enfoque metodológico y aplicaciones prácticas en la evaluación de medicamentos genéricos. Rev Med Urug 2000; 16(2):133-43.
15. Decreto N° 12/2007. Recomendaciones técnicas para la realización de estudios de bioquivalencia. Montevideo, Uruguay 12 de enero de 2007. Disponible en: http://archivo.presidencia.gub.uy/_Web/decretos/2007/01/266_15%2009%202006_00001.PDF [Consulta: 11 de jul de 2012].
16. United States Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation Research. Guidance for industry: Food effect on bioavailability and fed bioequivalence studies. Washington DC: 2002. Diponible en : http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf [Consulta:11 de jul de 2012)
17. Disposición 1746/2007 - ANMAT. Sustitúyese el Anexo I de la disposición 5040/2006, referido al Régimen de Buenas Prácticas para la Realización de Estudios de Biodisponibilidad/Bioequivalencia.. Buenos Aires, Argentina 2007.
18. Decreto Nro. 379/008. Investigación en seres humanos: aspectos éticos de la investigación que involucra seres humanos. Montevideo, Uruguay 4 de Agosto de 2008. Disponible en: http://archivo.presidencia.gub.uy/_web/decretos/2008/ 08/CM515_26%2006%202008_00001.PDF [Consulta: 11 de jul de 2012].
19. Decreto N° 261/009. Reglamentación de los centros y de los estudios de biodisponibilidad comparativa y/o bioequivalenica "in vivo" de medicamentos. Montevideo, Uruguay 1 de Junio de 2009. Disponible en: http://archivo.presidencia.gub.uy/_web/decretos/2009/06/ASUNTO74.pdf [Consulta: 11 de jul de 2012].
20. Decreto N° 369/010. Referencias para la realización de estudios de biodisponibilidad y bioequivalencia de medicamentos. Montevideo, Uruguay 13 de diciembre de 2010. Disponible en: http://archivo.presidencia.gub.uy/sci/decretos/2010/ 12/msp_97.pdf [Consulta: 11 de jul de 2011].
21. Decreto N° 097/011. Recomendaciones técnicas para la realización de estudios de equivalencia biofarmacéutica en centros especializados debidamente habilitados. Montevideo, Uruguay 2 de marzo de 2011. Disponible en: http://archivo.presidencia.gub.uy/sci/decretos/2011/03/msp_197.pdf [Consulta: 11 de jul de 2012].
22. Parrillo-Campiglia S, Cedrés M, Umpierrez O, Rodríguez P, Márquez S, Guarneri C, et al. Bioequivalence of two film-coated tablets of imatinib mesylate 400 mg: a randomized, open-label, single-dose, fasting , two-period , two-sequence crossover comparison in healthy male South American volunteers. Clin Ther 2009; 31(10):2224-32.
23. Estévez Carrizo FE, Parrillo S, Cedres M, Estévez Parrillo FT, Rodríguez P. Comparative bioavailability of two oral formulations of mycophenolate mofetil in healthy adult Uruguayan subjects: a case of highly variable rate of drug absorption. Int J Clin Pharmacol Ther 2010; 48(9):621-7.
24. Estévez Carrizo FE, Parrillo S, Cedrés M, Estévez Parrillo FT. Single dose relative bioavailability of a new quetiapine fumarate extended release formulation: a postprandial, randomized, open- label, two-way, crossover study in healthy Uruguayan volunteers. Clin Ther 2011; 33(6):738-45.
25. Estévez Carrizo FE, Ruíz S, Bellocq B, Leal C, Siri MT, del Campo MJ. Simultaneous itraconazole bioequivalence assessment and CYP3A phenotyping in Southamerican subjects. Int J Clin Pharmacol Ther 2005; 43(2):109-16.
26. Endrenyi L, Fritsch S, Yan W. Cmax/AUC is a clearer measure than Cmax for absorption rates in investigation of bioequivalence . Int J Clin Pharmacol Ther Toxicol 1991; 29(10):394-9.
27. Lacey LF, Keene ON, Duquesnoy C, Bye A. Evaluation of different indirect measures of rate of drug absorption in comparative pharmacokinetic studies. J Pharm Sci 1994; 83(2):212-15.
28. Schall R, Luus HG, Steinijans VW, Hauschke D. Choice of characteristics and their bioequivalence ranges for the comparison of absorption rates of immediate-release drug formulations. Int J Clin Pharmacol Ther 1994; 32(7):323-8.
29. Estévez Carrizo F, Giusti M, Parrillo S. Dextromethorphan O-demethylation genetic polymorphism in a hybrid South American population. Clin Pharmacol Ther 1999; 65(2):166.
30. Estevez F, Giusti M, Parrillo S, Oxandabarat J. Dextromethorphan O-demethylation polymorphism in the Uruguayan population. Eur J Clin Pharmacol 1997; 52(5):417-8.


{kind=link}
{kind=link}