










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkRevista Médica del Uruguay
versão On-line ISSN 1688-0390
Rev. Méd. Urug. vol.20 no.1 Montevideo mar. 2004
Experiencia metodológica en pesquisa neonatal de hiperfenilalaninemias.
Instituto de Genética Médica (Hospital Italiano)
Lic. María Jesús Roselli*, Dres. Aída Lemes†, Soraya Reyno‡,
Alicia Vaglio§, Roberto Quadrelli¶
Instituto de Genética Médica. Hospital Italiano. Montevideo, Uruguay
Resumen
La detección neonatal de errores congénitos del metabolismo mediante pesquisa neonatal consiste en la búsqueda sistemática en el recién nacido de aquellas afecciones para las que existe un tratamiento probadamente efectivo ante su instalación precoz. En el Instituto de Genética Médica del Hospital Italiano se realiza, desde noviembre de 1993, la cuantificación de fenilalanina sanguínea en el recién nacido en sangre seca sobre papel de filtro mediante método fluorométrico como test de pesquisa para hiperfenilalaninemias. Los reactivos son preparados en el laboratorio y la exactitud del método es controlada a través de un programa de control de calidad externo por método de referencia. En el presente trabajo se estimó el valor de punto de corte en función del cual considerar un resultado como presuntamente positivo para nuestra población (n=190), obteniéndose un valor de 2,5 mg/dl de fenilalanina. Se evaluó el método fluorométrico utilizado. El análisis de regresión entre los resultados obtenidos por el método en uso y el de referencia mostró que existe asociación lineal entre estos resultados (n=93; r=0,94; y= 0,87x + 0,519) y que aunque ambos métodos no son intercambiables, sí son equivalentes, lo cual otorga confianza al método en uso. Se examinó la estabilidad de la Phe en muestras de sangre recogidas sobre papel de filtro almacenadas a 4ºC. El porcentaje del aminoácido recuperado después de cinco años de almacenamiento (83% a 100%) y el test estadístico aplicado mostraron que el aminoácido tiene buena estabilidad almacenando las muestras en esas condiciones.
Palabras clave: FENILCETONURIAS-diagnóstico. FENILALANINA-análisis.
FLUOROMETRIA-métodos.
* Licenciada en Bioquímica. Facultad de Ciencias.
† Médico. Área de Errores Innatos del Metabolismo.
‡ Médico. Especialidad Genética Médica. Área de Dismorfología Clínica.
§ Médico. Especialidad Genética Médica y Pediatra. Subdirector.
¶ Médico. Especialidad Genética Médica. Director, Instituto de Genética Médica. Hospital Italiano.
Correspondencia: Lic. María Jesús Roselli
Br. José Batlle y Ordóñez 1647/309. Montevideo, Uruguay.
Instituto de Genética Médica. Br. Artigas 1632.
E-mail: mariroselli@hotmail.com
Recibido: 25/11/02.
Aceptado: 20/2/04.
Introducción
La detección neonatal de errores congénitos del metabolismo mediante pesquisa neonatal (PN) consiste en la búsqueda sistemática en el recién nacido de aquellas afecciones para las que existe un tratamiento probadamente efectivo si el mismo se comienza en forma precoz. En los países desarrollados, la PN forma parte esencial de los programas de prevención de la salud; las estrategias con que estos se ejecutan varían en los diferentes países, pero todos plantean un objetivo común: la identificación y prevención de desórdenes de la salud que puedan conducir a problemas potencialmente catastróficos. De esta manera, hablar de PN es equivalente a hablar de medicina preventiva, constituyendo una de las más eficaces herramientas que ha desarrollado la profilaxis pediátrica moderna(1-3).
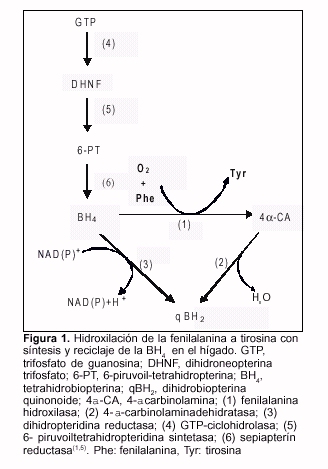
Entre las afecciones más comúnmente detectadas por PN se encuentran las hiperfenilalaninemias (HFA). Las HFA constituyen un grupo de enfermedades genéticas de herencia autosómica recesiva que se caracterizan por concentración plasmática del aminoácido esencial fenilalanina (Phe) persistentemente elevada. La base bioquímica de estas enfermedades es un trastorno en la reacción de hidroxilación de la Phe a tirosina. Este trastorno puede producirse por deficiencia primaria de la enzima hepática fenilalanina hidroxilasa (E.C. 1.14.16.1) (98% de los casos) o por deficiencia en la síntesis o reciclaje de su cofactor, la tetrahidrobiopterina (BH4), con la consecuente acumulación del aminoácido y sus derivados metabólicos en los fluidos corporales(1,4,5) (figura 1). Si bien existe considerable variación en el genotipo y el fenotipo, esta perturbación de la homeostasis metabólica va a tener consecuencias clínicas diversas entre las que se encuentran la deficiencia irreversible del desarrollo y funciones cognitivas, convulsiones y alteraciones epidérmicas(1,6-8).
La gran mayoría de los sujetos afectados están en alto riesgo de una incapacidad intelectual y neurológica que puede ser prevenida mediante la introducción de un tratamiento estrictamente controlado en las primeras semanas de vida(9,10). El tratamiento requiere la restauración del nivel de Phe sanguínea a valores tan cercanos al normal como sea posible, lo más precozmente posible y muy probablemente por el resto de la vida del paciente(1). Por tratarse de un aminoácido esencial (no es sintetizado por el organismo) esto se facilita limitando su ingesta, sin descuidar el aporte necesario para el correcto crecimiento y desarrollo del paciente(9-12).
En nuestro país no existirían aún estudios que permitan aportar datos sobre la incidencia de HFA(13), si bien es conocido que en caucásicos las HFA persistentes tienen una incidencia variable que se estima entre 1:4.000 a 1:14.000 recién nacidos(1,5).
Actualmente existen diferentes métodos para la detección de HFA que determinan la concentración de Phe en muestras de sangre sobre papel de filtro, plasma o suero: el método de inhibición bacteriana de Gutrhie (BIA)(14), diferentes procedimientos fluorométricos, basados en el método fluorométrico original de McCaman y Robins(15), métodos enzimáticos(16) y la espectrometría de masa en tándem(17).
En el Instituto de Genética Médica del Hospital Italiano se realiza, desde noviembre de 1993, la cuantificación de Phe sanguínea en el recién nacido en sangre seca sobre papel de filtro mediante método fluorométrico como test de pesquisa para HFA. Los reactivos utilizados son preparados en el laboratorio. La exactitud del método es controlada, desde la misma fecha, por un programa de control de calidad externo (PCCE) realizado por el Interlaboratory Quality Assurance for Newborn Screening (21494 Geesthach, Alemania) mediante método de referencia: cromatografía líquida de alta performance (HPLC). Se ha utlizado el valor 3,0 mg/dl de Phe sanguínea como punto de corte a partir del cual considerar un resultado presuntamente positivo de acuerdo a la bibliografía(18). Las muestras, ya sean de pesquisa o del PCCE, se almacenan a 4ºC con desecador hasta el momento de realizar el test.
Los objetivos del presente trabajo fueron:
- A partir de los datos de los recién nacidos evaluados, estimar el valor de punto de corte a partir del cual considerar un resultado presuntamente positivo para nuestra población.
- Evaluar el método fluorométrico en uso mediante el análisis de los resultados del PCCE.
- Examinar la estabilidad de la Phe en sangre seca sobre papel de filtro almacenada a 4ºC con desecador.
Material y método
Material de pesquisa neonatal
El test de pesquisa se realizó en recién nacidos entre los 2 días y 10 días de vida siempre que hubiesen recibido al menos 48 horas de alimentación proteica. La muestra de sangre se obtuvo por punción del talón depositando la misma en tarjetas de papel de filtro Schleicher & Schuell #903 (S & S #903) y una vez seca se almacenó a 4ºC con desecador hasta el momento de realizar el test.
Material del programa de control de calidad externo
El PCCE es realizado por el Interlaboratory Quality Assurance for Newborn Screening (21494 Geesthach, Alemania), mediante método de referencia (HPLC). Las muestras del PCCE se reciben cada dos meses, realizándose seis controles al año. Cada control consiste en diez tarjetas de papel de filtro con muestras de sangre de diferentes concentraciones de Phe (n=10). Junto con cada nueva serie de muestras a analizar se recibe también el resultado del control anterior en el que se informa, para las muestras que fueron positivas, la concentración de Phe en plasma obtenido por HPLC. Este valor es considerado como valor verdadero y es utilizado como referencia por los laboratorios participantes del PCCE al momento de examinar sus resultados(19).
Procedimiento empleado para determinar la concentración de Phe en sangre sobre papel de filtro en muestras de pesquisa y del PCCE
El procedimiento en uso se basa en el de Philips y de Boton(20) basado en el método fluorométrico de McCaman y Robins(15). El fundamento del mismo es la reacción de la Phe extraída de la muestra por acción del metanol, con ninhidrina y el dipéptido L-leucyl-L-alanina en condiciones óptimas de pH y temperatura para formar un complejo fluorescente. Este complejo es estabilizado por la adición de iones cobre. La intensidad de la fluorescencia emitida es proporcional a la concentración de Phe presente en la muestra(20). Todos los reactivos utilizados son preparados en el laboratorio, excepto los estándares de Phe (tiras de referencia para PKU, Sigma Diagnostics Co.).
Interpretación de los resultados de pesquisa neonatal
Se utilizó como punto de corte a partir del cual se considera un resultado presuntamente positivo el valor 3,0 mg/dl de Phe sanguínea, de acuerdo a la bibliografía(18).
Análisis estadístico de resultados
Para el análisis estadístico de los resultados se utilizó el software STATISTICA for Windows, release 4.5, Statsoft Inc. 1993.
Estimación del punto de corte
Con el fin de estimar el punto de corte se realizó el estudio de distribución de la variable: concentración de Phe sanguínea en el recién nacido, tomándose para ello todos los resultados obtenidos hasta ese momento que cumplieran los requisitos especificados en material de pesquisa neonatal (n=190). En función de este resultado se estimó el valor de punto de corte como la media más dos desvíos estándares (x + 2 SD).
Evaluación del método fluorométrico en uso. Análisis
de los resultados del PCCE
Se realizó un análisis de regresión lineal para los resultados positivos obtenidos en los 29 ensayos del PCCE realizados (n=93); la variable independiente (x) fue la concentración de Phe en plasma determinada por HPLC y la variable dependiente (y) fue el resultado obtenido en papel de filtro por el procedimiento flurométrico en uso. Se realizó un test de Student para la ordenada en el origen (a=0) y para la pendiente de la recta (b=1), a fin de establecer si existe diferencia constante o proporcional, o ambas, entre el método en uso y el de referencia (p=0,05).
Estudio de la estabilidad de la Phe en sangre sobre papel de filtro
Se examinó la estabilidad del aminoácido en muestras positivas del PCCE guardadas a 4ºC con desecador después de cinco años de almacenamiento (n=17). Se determinó el porcentaje del aminoácido retenido en cada muestra y se realizó el test de Student para grupos pareados a fin de establecer si existían diferencias entre la concentración de Phe inicial y la obtenida después de dicho período de tiempo (p=0,05).
Resultados
Estimación del punto de corte
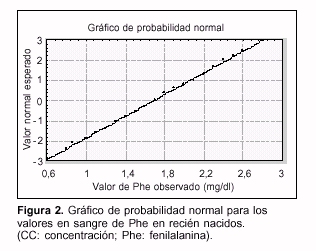
- El análisis estadístico de los valores de la concentración de Phe sanguínea en el recién nacido mostró que esta variable se encuentra normalmente distribuida, como se puede apreciar en la figura 2, en la que se encuentran graficados los valores esperados en términos de valor z para una distribución normal en función de los valores de Phe obtenidos.
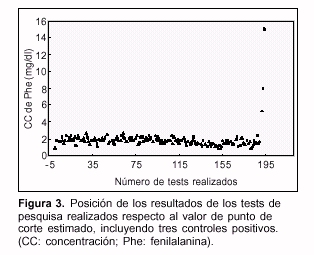
- El valor medio de Phe (n = 190) fue: x = 1,7 ± 0,4 mg/dl (x ± SD), obteniéndose un valor de punto de corte de 2,5 mg/dl (x + 2 SD). En la figura 3 se muestran los resultados de los tests de pesquisa realizados respecto al valor de punto de corte; se incluyeron también tres controles positivos.
Evaluación del método fluorométrico en uso. Análisis
de los resultados del PCCE
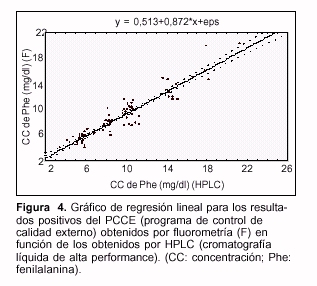
- El análisis de regresión mostró que existe asociación lineal entre los resultados obtenidos por ambos métodos (n=93; r = 0,94; p=0,95) expresada por la ecuación de regresión y = 0,87x + 0,51 (y = bx + a) (figura 4). Se obtuvo un valor para la pendiente de la recta de 0,87 ± 0,03 (b ± SDb)y para la ordenada en el origen de 0,51± 0,32 mg/dl de Phe (a ± SDa).
- Los tests estadísticos realizados para la pendiente de la recta y para la ordenada en el origen mostraron que la pendiente de la recta es significativamente diferente del valor 1 y que la ordenada en el origen no lo es del valor 0 (p=0,05).
Estudio de la estabilidad de la Phe en sangre sobre papel de filtro
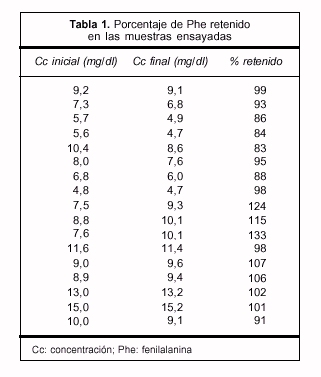
- Las muestras examinadas retuvieron entre 83% y 99% del aa presente originalmente. En algunas muestras, el porcentaje retenido fue mayor de 100%, ya que el valor actual fue ligeramente mayor que el inicial (tabla 1).
- El test de Student de grupos pareados no mostró diferencias significativas entre la concentración actual y la presente hace cinco años (p=0,05).
Discusión
Las HFA persistentes si no son detectadas en etapa preclínica (primeros días de vida) muy probablemente determinen incapacidad intelectual y neurológica irreversible. El tratamiento instituido en las primeras semanas de vida evita estas consecuencias. En un país donde la mortalidad infantil por causas como diarrea e infecciones ha sido superada, la implementación a nivel nacional de un programa de pesquisa para HFA en el recién nacido puede resultar sumamente beneficioso, permitiendo no sólo el desarrollo de personas normales si se instaura rápidamente el tratamiento en los casos positivos, sino también conocer la incidencia de las mismas en nuestro país. Se debe tener en cuenta que los estudios costo/beneficio realizados en los diferentes países donde se aplica la PN han justificado siempre su aplicación(1).
Estimación del punto de corte
En el presente trabajo se estimó el valor de punto de corte a partir de los resultados de pesquisa de nuestra población. El análisis estadístico de estos datos mostró que la variable concentración de Phe en la sangre del RN se encuentra normalmente distribuida. Dicho resultado está de acuerdo con lo encontrado por Holtzman y colaboradores(21) y coincidiría con lo expuesto por Siest en el 2º Congreso de Bioquímica Clínica(22.--
22). Siest señaló que, generalmente, los componentes de un sistema, como puede ser la sangre, que se encuentran al inicio de una vía metabólica y se utilizan como sustrato para la síntesis de otros metabolitos no poseen una variabilidad biológica elevada, encontrándose sus valores normalmente distribuidos. Por el contrario, los metabolitos que se excretan al final de una vía metabólica poseen una variabilidad biológica mayor y sus determinaciones no se distribuyen normalmente. La Phe es un aminoácido esencial que debe ser aportado por la dieta para un adecuado desarrollo del sujeto, es fuente por su oxidación de un aminoácido no esencial, la tirosina, a partir de la cual se sintetizan los neurotransmisores y la melanina. Estas características podrían justificar, de acuerdo a lo expuesto por Siest, que sus valores de concentración en la sangre tengan distribución normal.
El valor de punto de corte obtenido, 2,5 mg/dl de Phe, difirió ligeramente del usado hasta el momento en el Instituto de Genética Médica (3,0 mg/dl). Este valor puede considerarse válido ya que se obtuvo a partir de un número significativo de recién nacidos de nuestra población y disminuiría la posibilidad de falsos negativos, sin incrementar sensiblemente la de falsos positivos. Weiner y colaboradores(23) señalaron que la disminución del valor de punto de corte de >4 mg/dl a >2 mg/dl resultó en un aumento de la tasa de muestras adicionales (repetición de la toma) de 0,01% a sólo 0,03%, que representaría 30 requerimientos adicionales en 100.000 pesquisas realizadas. Es importante considerar el número de falsos positivos a la hora de solicitar nueva muestra, debido a la ansiedad que esto genera en los padres del recién nacido(24). Aunque este valor de punto de corte es ligeramente mayor que el recomendado por Doherty y colaboradores(25), 2,0 mg/dl, no existiría un elevado riesgo de no detectar un individuo positivo, ya que en el servicio la toma de la muestra se realiza a las 48 horas de vida como mínimo y siempre que el recién nacido haya recibido 48 horas de alimentación proteica. Por otra parte, Jew y colaboradores(26) demostraron que el método fluorométrico de McCaman y Robins(15) es capaz de identificar recién nacidos afectados a las 12 horas de vida. A partir del presente trabajo este valor ha sido (fue) adoptado como nuevo valor de punto de corte al realizar la PN.
Evaluación del método fluorométrico en uso. Análisis
de los resultados del PCCE
Existen diferentes métodos para detectar niveles en sangre de Phe aplicables como método de pesquisa para HFA en los primeros días de vida. Respecto al utilizado en el servicio, el presente trabajo muestra que existe asociación lineal entre los resultados obtenidos por éste y el de referencia. De acuerdo con los tests estadísticos realizados para la pendiente y la ordenada en el origen de la recta de regresión, no existe una diferencia constante entre este método y el de referencia, pero sí una diferencia proporcional que es el valor de la pendiente; por cada mg/dl determinado por HPLC, fluorométricamente se determinarán 0,87 mg/dl. De acuerdo con lo anterior se puede decir que ambos métodos no son intercambiables pero sí son equivalentes.
Se puede afirmar entonces que el método en uso es confiable a la hora de realizar el test de pesquisa; esto es importante no solamente en el momento de detectar un recién nacido potencialmente enfermo, sino también como parte del posterior seguimiento del paciente, a la hora de realizar los controles de Phe sanguínea una vez comenzado el tratamiento. Es de destacar también que, de acuerdo a los informes del PCCE, no se han registrado hasta el momento resultados falsos positivos o falsos negativos en los nueve años de control de calidad externo realizados. Es oportuno señalar que los tests basados en ensayos fluorométricos tienen una especificidad y sensibilidad mayores que el clásico ensayo de Gutrhie (BIA)(14), ampliamente difundido años atrás, ya que este último, al ser semicuantitativo, depende de la subjetividad del operador(1). Una revisión del programa de PN para HFA en el Reino Unido(27), encontró que todos los falsos negativos para PKU estuvieron asociados con tests realizados por BIA y ninguno estuvo asociado a tests realizados por fluorometría. La confiabilidad del método es significativa si se considera que no se utilizan presentaciones comerciales de reactivos (kit), sino que todos los reactivos son preparados en el laboratorio, lo que también disminuye los costos.
Estudio de la estabilidad de la Phe en sangre sobre papel de filtro
Los resultados indicaron que la Phe en sangre recolectada sobre papel de filtro tiene buena estabilidad después de cinco años de almacenamiento en las condiciones descritas. Este resultado concuerda con lo encontrado por Levy y colaboradores(28), en el que muestras guardadas a temperatura ambiente, con cinco años o menos de almacenamiento, retuvieron entre 40% y 100% de la Phe original.
En algunas muestras el porcentaje retenido fue mayor de 100%, ya que el valor actual fue ligeramente mayor que el obtenido originalmente. Al remitirse a estas muestras se observó que el valor de Phe original determinado por fluorometría era significativamente menor que el obtenido por HPLC, por lo que inicialmente habría ocurrido una subestimación del aminoácido presente en las mismas. Una buena estabilidad del aminoácido a 4ºC justificaría el almacenamiento de las muestras en papel durante (al menos) cinco años. De esta manera si es necesario remitirse a la muestra original, se tendrá un reflejo válido de la concentración de Phe en el momento en que se realizó el test; esto permitirá, por ejemplo, diferenciar un error de laboratorio de la posible variabilidad biológica en la concentración de Phe, una fuente de falsos negativos en los tests de pesquisa, especialmente en aquellas no PKU(1).
Conclusiones
- El valor de punto de corte obtenido para nuestra población fue 2,5 mg/dl de Phe, el cual al ser ligeramente menor que el usado actualmente disminuirá la posibilidad de obtener un resultado falso negativo, sin incrementar significativamente el número de falsos positivos.
- Existe asociación lineal (r = 0,94; p = 0,95) entre el método fluorométrico usado y el método de referencia y si bien ambos métodos no son intercambiables, sí son equivalentes.
- La Phe en muestras de sangre recogida sobre papel de filtro almacenado a 4ºC con desecador es estable después de cinco años, por lo que dentro de este período de tiempo la concentración de Phe encontrada en la muestra será un reflejo válido de la presente originalmente.
Agradecimientos
Queremos agradecer a Luz Mery Bernal PhD, por las sugerencias realizadas en la corrección del presente artículo.
Summary
Newborn detection of metabolic congenital abnormalities by newborn screening consists of systematic searching of affections that could be treated in early stages. Since November 1993, quantification of phenylalanine was obtained by filter paper blood spots collected from newborns by fluorometric technique for hyperphenylalanine. Reagents were prepared in the laboratory, accuracy of the method was controlled by an external quality control program (reference method).
Cutoff for the population (n=190) was 2.5 mg/dl of phe-nylalanine. The fluorometric technique used was assessed. Regression analysis for the results obtained by fluorometric technique and the reference method showed lineal association among those techniques (n=93; r=0.94; y=0.87x + 0.519) and reliance on the proposed technique based on the equivalence of the techniques. Phe stability was analysed using filter paper blood spots storaged at 4°C. Amino acid profiles retrieved after five years of storage (83% to 100%) and the statistic test used showed that the amino acid is stable when storaged under these conditions.
Résumé
La découverte néonatale d'erreurs congénitales du méta-bolisme au moyen de recherche néonatale, comprend la recherche systhématique chez le nouveau-né des affec-tions pour lesquelles il existe un traitement prouvé effectif en tant que précoce. À l'institut de Génétique Médicale de l'Hôpital Italien on réalise, depuis novembre 1993, la quantification de phénylalaline sanguine chez le nouveau-né en sang sec sur papier filtre avec une méthode fluoro-métrique comme test de recherche pour hyperphénylala-linémies. Les réactifs sont préparés au laboratoire et l'exactitude de la méthode est évaluée au moyen d'un programme de contrôle de qualité externe par méthode de référence. Dans ce travail, on a estimé la valeur de point de coupe en fonction duquel considérer un résultat comme positif pour notre population (n=190), obtenant une valeur de 2,5 mg/dl de phénylalaline. On a évalué la méthode fluorométrique utilisée. L'analyse de régression entre les résultats obtenus par la méthode utilisée et celui de référence a montré qu'il existe une relation linéaire entre ces résultats (n=93 ; r=0,94 ; y= 0,87x + 0,159) et même si les deux méthodes ne sont pas échangeables elles sont équivalentes, ce qui donne confiabilté à la méthode utilisée. On a examiné la stabilité de la Phe dans des échantillons de sang recueillis sur papier de filtre à 4ºC. Le pourcentage de l'aminoacide récupéré après cinq ans de stockage (83% à 100%) et le test statistique appliqué ont montré que l'aminoacide a une bonne stabilité si on stocke les échantillons dans ces conditions.
Bibliografía
1. Scriver CR, Kaufman S. Hyperphenylalaninemia: Phenylalanine hydroxilase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: Mc Graw Hill, 2001: 1667-724.
2. Committee on Genetics. Aspectos del screening neonatal. Pediatrics (ed. esp.) 1992; 33: 103-7.
3. Bickel H. Rationale of Neonatal Screening for Inborn Errors of Metabolism. In: Bickel H, Guthrie R, Hammersen G, eds. Neonatal Screening for Inborn Errors of Metabolism. Berlín: Springer, 1980: 1-6.
4. Wilcox WR, Cederbaum SD. Aminoacid metabolism. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, eds. Emery and Rimoin's: Principles and Practice of Medical Genetics. 4ª ed. London: Churchill Livingstone, 2002: 2405-40.
5. Smith I, Lee P. The hyperphenylalaninemias. In: Fernandes J, Saudubray JM, van der Berghe GV, eds. Inborn Metabolic Diseases. Diagnosis and treatment. 3ª ed. Berlín: Springer-Verlag, 2000: 170-83.
6. Corsello G, Bosco P, Calí F, Greco D, Cammarata M, Caccio M, et al. Maternal phenylketonuria in two Sicilian families identified by maternal blood phenylalanine level screening and identification of a new phenylalanine hydroxylase gene mutation (P470L) Eur J Pediatr 1999; 158(1): 83-4.
7. Enns GM, Martinez DR, Kuzmin AI, Koch R, Wakeem CK, Woo SL, et al. Molecular correlations in phenylketonuria: mutation patterns and corresponding biochemical and clinical phenotypes in a heterogeneous California population. Pediatr Res 1999; 46(5): 594-602.
8. Guttler F, Azen C, Guldberg P, Romstad A, Hanley W, Levy HL. Relationship among genotype, biochemical phenotype, and cognitive performance in females with phenylalanine hidroxylase deficiency: report from the maternal phenylketonuria collaborative study. Pediatrics 1999; 104(2 Pt 1): 258-62.
9. Smith I. Treatment of phenylalanine hydroxylase deficiency. Acta Paediatr Suppl 1994; 407: 60-5.
10. Schmidt H, Burgard P, Pietz J, Rupp A. Intelligence and professional career in young adults treated early for phenylketonuria. Eur J Pediatr 1996; 155(Suppl 1): S97-100.
11. Cockburn F, Clark BJ. Recommendations for protein and amino acid intake in phenylketonuric patients. Eur J Pediatr 1996; 155(Suppl 1): S125-9.
12. Azen C, Koch R, Friedman E, Wenz E, Fishler K. Summary or findings from the United States Collaborative Study of children treated for phenylketonuria. Eur J Pediatr 1996; 155(Suppl 1): S29-32.
13. Rafaelli A, Dell' Acqua A. Pesquisaje neonatal (PN) de hiperfenilalaninemias (HFA): 1er. caso detectado en el Uruguay. Congreso Uruguayo de Bioquímica Clínica, 1º. Montevideo. Uruguay, 1997: 71-2.
14. Gutrhie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large population of newborn infants. Pediatrics 1963; 32: 338-43.
15. McCaman MW, Robins E. Fluorometric method for the determination of Phenylalanine in serum. J Lab Clin Med 1962; 59: 885-90.
16. Wendel U, Koppelkaumm M, Hummel W, Sander J, Langenbeck U. A new approach to the newborn screening for hyperphenylalaninemia use of L- pheylalnine dehydrogenase and microtiter plates. Clin Chim Acta 1990; 192(3): 165-70.
17. Chace DH, Millington DS, Terada N, Kahler SG, Roe CR, Hofman LF. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by Tandem Mass Spectrometry. Clin Chem 1993; 39(1): 66-71.
18. Chamoles NA, Spécola N. Encefalopatías evolutivas: errores congénitos del metabolismo. In: Fejermann N y Fernández Álvarez E, eds. Neruología Pediátrica. 2ª ed. Buenos Aires: Panamericana, 1997: 319-82.
19. Mathias D, Böttcher M, Gebhard J. Quality Control in Neonatal Screening for Inborn Errors of Metabolism. Short communication. Clin Lab 1998; 44: 51-60.
20. Philips RE, de Boton R. (A Clinical Laboratory Procedure from G.K. Turner Associates, Inc., 2524 Pulgas Avenue, Palo Alto, California 94303): A simplified determination of blood phenylalanine using samples collected on paper.
21. Holtzman NA, Mc Cabe ERG, Cunningham GC, Berry HK. Screening for phenylketonuria. N Engl J Med 1981; 304(21): 1300-1.
22. Siest G. Simposio Valores de Referencia. Congreso de Bioquímica Clínica, 2º; Montevideo, Uruguay; 1999: 23-5.
23. Weiner DL, Canton RM, Mitchel PL, Mannes P. False positive rate in neonatal phenylketonuria (PKU) screening using a 2 mg/dl phenylalanine (PA) cut-off. Am J Hum Genet 1981; 33: 94 A.
24. Erbe RW, Levy HL. Screening neonatal. In: Rimoin DL, Connor JM, Pyeritz RE, eds. Emery and Rimoin's: Principles and Practice of Medical Genetic. 3ª ed. New York: Churchill Livingstone, 1997: 581-93.
25. Doherty LB, Rohr FJ, Levy HL. Detection of phenylketonuria in the very early newborn blood specimen. Pediatrics 1991; 87(2): 240-4.
26. Jew K, Kan K, Koch R, Cunningham GC. Validity of screening early collected newborn specimens for phenylketonuria using a fluorometric method. Screening 1994; 3: 1-9.
27. Smith I, Cook B, Beasley M. Review of neonatal screening programme for phenylketonuria. BMJ 1991; 303(6798): 333-5.
28. Levy HL, Simmons JR, MacCready RA. Stability of amino acids and galactose in the newborn screening filter paper blood specimen. J Pediatr 1985; 107(5): 757-9.

