










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkRevista Médica del Uruguay
versão On-line ISSN 1688-0390
Rev. Méd. Urug. vol.19 no.2 Montevideo ago. 2003
Aportes de la microscopía electrónica de transmisión al diagnóstico de la disquinesia ciliar
Dra. M. Mónica Brauer1, Br. Lorena Viettro2
Laboratorio de Biología Celular. Instituto de Investigaciones Biológicas Clemente Estable
Resumen
La disquinesia ciliar primaria es una enfermedad congénita que aúna un grupo heterogéneo de condiciones patológicas que se caracterizan por la presencia de alteraciones microanatómicas o funcionales, o ambas, en las cilias y los flagelos espermáticos. Las manifestaciones clínicas de este desorden son variadas y se caracterizan por la aparición temprana de infección recurrente de las vías auditivo-respiratorias. Además de los determinantes genéticos, la propia infección e inflamación recurrente -así como algunos agentes ambientales- pueden provocar alteraciones ciliares. Estas alteraciones son, sin embargo, de tipo inespecífico y conducen a la llamada disquinesia ciliar secundaria. En el presente trabajo se describen los resultados obtenidos del estudio de 40 biopsias de mucosa nasal y bronquial provenientes de 33 pacientes sospechosos de portar alguna de estas formas de disquinesia ciliar, las cuales fueron remitidas por los médicos tratantes al Laboratorio de Biología Celular del Instituto de Investigaciones Biológicas Clemente Estable para su estudio por microscopía electrónica de transmisión. En su conjunto estos estudios mostraron que 9 de los pacientes (27%) presentaban alteraciones ultraestructurales ciliares compatibles con el diagnóstico de disquinesia ciliar primaria. Tres de ellos tenían situs inversus totalis. El 52% de los pacientes presentó alteraciones ciliares secundarias. A pesar de sus manifestaciones clínicas, 21% de los pacientes restantes mostró una ultraestructura ciliar normal. En su conjunto, los resultados obtenidos en la población estudiada apuntan a confirmar la alta frecuencia con que se presentan las alteraciones ciliares en los pacientes respiratorios crónicos. Teniendo en cuenta que el diagnóstico precoz de esta enfermedad es esencial para prevenir el desarrollo de lesiones respiratorias irreversibles, se destaca la importancia de indicar el examen ultraestructural de las cilias en los pacientes portadores de situs inversus y sus hermanos, así como en los individuos afectados de enfermedad respiratoria crónica severa de inicio precoz y carentes de diagnóstico etiológico.
Palabras clave: ENFERMEDADES RESPIRATORIAS.
SÍNDROME DE KARTAGENER - diagnóstico.
MICROSCOPÍA ELECTRÓNICA.
TRASTORNOS DE LA MOTILIDAD CILIAR - diagnóstico.
1. PhD. Jefe del Laboratorio de Biología Celular, Instituto de Investigaciones Biológicas Clemente Estable (IIBCE).
2. Becaria del Laboratorio de Biología Celular, IIBCE
Correspondencia: Dra. M. M. Brauer
Laboratorio de Biología Celular, IIBCE
Avenida Italia 3318, 11600 Montevideo - Uruguay
E-mail: brauer@iibce.edu.uy
Recibido: 24/9/02.
Aceptado: 11/7/03.
Introducción
El batido ciliar constituye una parte importante del mecanismo de defensa de las vías respiratorias y cumple la importante tarea de eliminar partículas y microorganismos inhalados, así como las secreciones producidas localmente. El sistema mucociliar está constituido por la capa de moco y el estrato de cilias subyacente. Para que el transporte mucociliar sea eficiente es necesario que el moco posea una viscosidad y elasticidad adecuadas, y que las cilias se muevan apropiadamente. Los desórdenes congénitos del transporte mucociliar pueden deberse a alteraciones estructurales o funcionales, o ambas, de la cilias (disquinesia ciliar primaria) o a alteraciones en la viscosidad de las secreciones bronquiales (fibrosis quística)(1,2). El transporte mucociliar deficitario impide que los agentes patógenos sean eliminados de las vías respiratorias, favoreciendo así la recurrencia de la infección respiratoria(1,2).
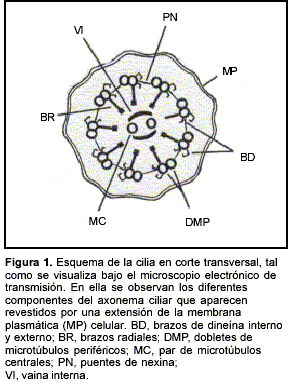
En 1975, se presentan los primeros casos que relacionaban la esterilidad masculina con alteraciones en el flagelo espermático(3,4). Estos estudios pioneros mostraron que estos pacientes, además de ser infértiles, poseían una historia de infecciones auditivas y respiratorias recurrentes, y en algunos casos presentaban las manifestaciones clínicas de la denominada tríada de Kartagener (sinusitis, bronquiectasias y situs inversus). Estudios posteriores demostraron que estos individuos tenían un transporte mucociliar deficitario(5) y que sus cilias (figura 1) mostraban alteraciones microanatómicas que sólo eran visibles bajo el microscopio electrónico de transmisión(6,7).
En 1977, Eliasson y colaboradores(8) denominaron como síndrome de inmovilidad ciliar al defecto congénito de cilias y flagelos causante de la infección recurrente de las vías auditivo-respiratorias y la infertilidad masculina. Actualmente, este nombre ha sido cambiado por el de síndrome de disquinesia ciliar, al comprobarse que en algunos casos las cilias defectuosas son capaces de moverse, aunque lo hacen en una forma no coordinada o disquinética(9,10). Así, el síndrome de disquinesia ciliar primario se define actualmente como una enfermedad de origen genético, generalmente autosómico y recesivo(11,12) que agrupa una variedad heterogénea de condiciones patológicas caracterizadas por alteraciones funcionales o estructurales, o ambas, en las cilias(2,13,14). Clásicamente se estimó que esta enfermedad afectaba a uno de entre 15.000 y 30.000 individuos, pero actualmente se cree que su prevalencia está subestimada -aun en los países desarrollados- y que existe un alto número de pacientes no diagnosticados(2,14,15). Esto se debe a que, si bien los primeros síntomas de la enfermedad se presentan durante el período neonatal, existen variaciones en la forma y edad de presentación, por lo que su diagnóstico se ve frecuentemente postergado(12).
Además de los determinantes genéticos, existen factores ambientales que pueden alterar a las cilias conduciendo a la llamada disquinesia ciliar secundaria(16,17). Estas alteraciones secundarias son causadas por las propias infecciones bacterianas o virales, por el micoplasma y por agentes ambientales irritantes, tales como el humo del tabaco y la polución ambiental. Las mismas pueden aparecer en forma aislada o asociadas con las determinadas genéticamente, llegando en algunos casos a afectar la integridad del epitelio respiratorio.
El diagnóstico precoz de la disquinesia ciliar primaria ha mostrado ser crucial para disminuir la morbimortalidad de estos pacientes y mejorar sustancialmente su calidad de vida(2,14,18,19). Actualmente se acepta que el tratamiento de esta enfermedad requiere de una rigurosa fisioterapia pulmonar, antibioticoterapia profiláctica o patógeno-específica e inmunización contra los patógenos respiratorios comunes en la comunidad(18,19). Los estadios tardíos de la enfermedad pueden llegar a requerir cirugía para eliminar las bronquiectasias, llegándose al trasplante pulmonar en los casos terminales(1,20).
En el presente trabajo se describen los resultados obtenidos del estudio de 40 biopsias de mucosa respiratoria de cornete inferior y bronquio provenientes de 33 pacientes portadores de enfermedades auditivo-respiratorias crónicas, carentes de diagnóstico etiológico, que fueron remitidas por los neumólogos y otorrinolaringólogos tratantes al Laboratorio de Biología Celular del Instituto de Investigaciones Biológicas Clemente Estable (IIBCE) para la realización de estudios de ultraestructura ciliar. Se discute la utilidad de la microscopía electrónica como herramienta para el diagnóstico diferencial de la disquinesia ciliar primaria y secundaria, y se analiza la importancia de implementar métodos complementarios para caracterizar plenamente estas enfermedades.
Material y método
El presente estudio fue realizado sobre 40 biopsias de mucosa respiratoria de cornete inferior (n=32) y bronquio (n=7) que fueran remitidas al Laboratorio de Biología Celular del IIBCE para la realización de estudios de ultraestructura ciliar. Estas biopsias provinieron de una población de 33 pacientes pertenecientes a diferentes centros asistenciales públicos y privados de Uruguay. Veinticuatro de los pacientes estudiados fueron niños de entre 0 y 15 años (12 niños y 12 niñas) y 9 adultos de entre 16 y 62 años (2 del sexo masculino y 7 del femenino). Todos los pacientes carecían de diagnóstico etiológico luego de estudios exhaustivos que descartaron otras enfermedades que provocan cuadros clínicos similares, tales como la fibrosis quística(21), los desórdenes inmunológicos y las deficiencias congénitas de ?1-antitripsina.
Las biopsias fueron obtenidas por los neumólogos u otorrinolaringólogos tratantes bajo anestesia general y en block quirúrgico. Los médicos tratantes obtuvieron el consentimiento informado de los pacientes adultos y de los padres o tutores de los pacientes menores de edad, siguiendo las normativas propias de cada institución. Con el fin de mejorar la integridad epitelial, las biopsias fueron realizadas por lo menos a tres semanas del último episodio infeccioso o alérgico, en algunos casos acompañado de antibioticoterapia indicada por el médico tratante. Se realizaron biopsias de cornete inferior en 32 de los pacientes y en siete de ellos se efectuaron, simultánea o posteriormente, biopsias de mucosa bronquial. Una de las biopsias bronquiales fue obtenida durante una lobectomía. Las mucosas respiratorias fueron inmediatamente fijadas en glutaraldehído al 2,5% en tampón fosfato 0,1M (pH: 7,2-7,4) y trasladadas en este fijador a 4ºC al Laboratorio de Biología Celular del IIBCE, donde fueron procesadas dentro de las 48 horas siguientes a su arribo.
Bajo el microscopio de disección se procedió a localizar la superficie epitelial y se fragmentaron las biopsias en trozos más pequeños (aproximadamente 0,5 x 0,5 x 1 mm). Algunos fragmentos se reservaron como respaldo en el líquido fijador a 4ºC y los fragmentos a estudiar se procesaron de acuerdo con técnicas convencionales de preparación de materiales para su estudio por microscopía electrónica de transmisión. Esto incluyó: 1) lavado en tampón fosfato 0,1M (pH: 7,2-7,4) por una hora; 2) posfijación en tetróxido de osmio al 1% en tampón fosfato 0,1M por 90 minutos y al resguardo de la luz; 3) lavado en tampón fosfato 0,1M por una hora; 4) deshidratación en alcoholes de graduación creciente (25%, 50%, 70%, 90% y 100%) (2 x 10 minutos cada uno) seguidos de tres lavados en acetona absoluta de 15 minutos cada uno; 5) inclusión en resina epoxi (Durcupan ACM, Fluka). Con el fin de mejorar la penetración de la resina, los tejidos permanecieron en una mezcla en partes iguales de Durcupan ACM y acetona absoluta por 12 horas a temperatura ambiente. Luego de dos pasajes de 3 horas por resina pura, los tejidos fueron incluidos en resina fresca y en bloques individuales debidamente identificados. Los bloques fueron polimerizados por 24 horas a 60ºC.
Los cortes fueron realizados utilizando un ultramicrótomo Porte Bloom MT1. En una primera etapa se realizaron cortes semifinos (0,25 - 0,5 mm de espesor) los cuales fueron montados sobre portaobjetos y teñidos con azul de toluidina borácico al 1%. Estos cortes fueron examinados bajo el microscopio de luz con el fin de localizar la superficie epitelial ciliada. Este procedimiento se repitió varias veces a distintas profundidades del mismo bloque de tejido y en los distintos bloques obtenidos de la toma biópsica de cada paciente. Las áreas epiteliales seleccionadas fueron talladas bajo la lupa del ultramicrótomo y se realizaron cortes ultrafinos (600-700 A de espesor) los cuales fueron montados en grillas de cobre, utilizando como soporte un film de Formvar (Fluka). Los cortes ultrafinos fueron contrastados con acetato de uranilo saturado (12-16 horas a temperatura ambiente y al resguardo de la luz), seguido de citrato de plomo al 0,06% (10 minutos en atmósfera de hidróxido de sodio). El examen ultraestructural fue realizado utilizando un microscopio de transmisión JEOL 100CX II, operado a 60 KV. Las imágenes de las cilias fueron fotografiadas en placas Kodak de 6,5 x 9 cm (SPI), reveladas en Kodak D11 y positivadas en papel fotográfico Ilford Multigrado. La ultraestructura ciliar fue evaluada sobre un mínimo de 40 y un máximo de 350 cilias en corte perfectamente transversal por paciente. Con el fin de mejorar la calidad de las imágenes electrónicas y en particular la visualización de los brazos de dineína, las microfotografías electrónicas fueron digitalizadas utilizando un escáner Genius ColorPage Vivid Plus (800 dpi) y posteriormente sometidas a ajustes de brillo y contraste utilizando el programa Adobe Photoshop. Las imágenes digitales fueron impresas en papel satinado Hewlett Packard HP Premium Inkjet utilizando una impresora Hewlett Packard DeskJet 840C.
Resultados y discusión
Todos los pacientes estudiados presentaron dos o más de los signos y síntomas que se muestran en la figura 2. La mayoría de ellos presentó un florido cuadro respiratorio, que incluyó neumonías bacterianas recidivantes, bronquiectasias (diagnosticadas por radiografía común y confirmadas por tomografía axial computarizada [TAC] de alta resolución), bronquitis, rinitis, otitis y asma. Catorce de los pacientes presentaron problemas auditivo-respiratorios antes del año de vida y cuatro de ellos presentaron situs inversus totalis (tabla 1).


Las alteraciones ciliares fueron clasificadas de acuerdo con la nomenclatura propuesta por Barlocco y colaboradores(22), con algunas modificaciones (tabla 2). Las alteraciones consideradas de origen genético o primario se encuadraron en los tipos I a IV, mientras que las afecciones consideradas como inespecíficas y secundarias a la propia infección e inflamación recurrente, se encuadraron en los tipos V y VI. La relación entre los datos clínicos de los pacientes y la presencia de estas alteraciones ciliares se muestra en la tabla 1, mientras que su incidencia en la población estudiada se resume en la figura 3. En su conjunto el examen ultraestructural de las cilias permitió discriminar tres grupos de pacientes: a) portadores de lesiones ciliares compatibles con el síndrome de disquinesia ciliar primario (27%); b) portadores de lesiones ciliares secundarias o inespecíficas (52%) y c) individuos con ultraestructura ciliar normal (21%).


Dentro del primer grupo, que incluyó a nueve de los pacientes, cinco mostraron deficiencias en el número de brazos de dineína, proteína con actividad ATPasa responsable del movimiento ciliar (figuras 1, 4a y 4b). La deficiencia en los brazos de dineína fue la alteración inicialmente detectada en los primeros estudios de microscopía electrónica y la que se describe como la alteración primaria más frecuente(6). Estos estudios, sin embargo, se basaron en el empleo de métodos cualitativos para describir esta reducción en el número de brazos de dineína, lo que condujo en algunos casos a subestimar el número de pacientes afectados. Estudios más recientes han mostrado la importancia de utilizar métodos cuantitativos para diagnosticar esta alteración(14,15) y la aplicación de los mismos ha permitido revertir el diagnóstico de muchos pacientes inicialmente considerados normales(15). Aunque el número normal de brazos de dineína por cilia es de nueve brazos internos y nueve externos (figura 1), en la práctica existen limitaciones que impiden verlos simultáneamente. En particular, los brazos internos tienden a preservarse pobremente con las técnicas convencionales de microscopía electrónica(2). Si bien aún existen discrepancias respecto al número medio normal de brazos de dineína internos y externos por cilia que pueden observarse en un corte(14,15), en nuestros estudios se tomaron como normales los valores medios superiores a 3,5 brazos de dineína internos y 7,0 brazos de dineína externos por corte transversal de cilia. Estos valores deberán ser reevaluados en el futuro, cuando se alcance un consenso entre los diferentes grupos de trabajo del mundo.
Dos de los pacientes que presentaron afecciones ciliares compatibles con el diagnóstico de disquinesia ciliar primaria mostraron alteraciones en el número y distribución de los microtúbulos (disquinesia tipo III, tabla 2, figuras 1 y 4c), mientras que los dos restantes presentaron cilias aplásicas, carentes de axonema (disquinesia tipo IV, tabla 2, figura 4d). Estos resultados no son inesperados si se considera que el axonema ciliar incluye más de 200 proteínas(23), por lo que actualmente se acepta que existen alteraciones primarias que afectan a otros componentes de las cilias, tales como los microtúbulos, los brazos radiales, la vaina interna y los puentes de nexina(2). Estas alteraciones son capaces de provocar la disquinesia ciliar primaria e inducir, por lo tanto, cuadros clínicos similares.
Tres de los pacientes en los que se detectaron alteraciones ciliares primarias tenían situs inversus totalis. Cabe señalar, sin embargo, que dos de ellos -hermanos entre sí- presentaron una marcada reducción en el número de brazos de dineína, pero sólo uno de ellos presentó situs inversus. Estos resultados señalan que la disquinesia ciliar primaria no se acompaña "necesariamente" de situs inversus y confirman el concepto de que en ausencia de un batido ciliar eficiente durante el desarrollo embrionario, la distribución de los órganos impares queda librada al azar, dando 50% de individuos normales y 50% de individuos portadores de situs inversus(2).
Diecisiete de los 33 pacientes estudiados presentaron alteraciones ciliares de tipo inespecífico, consistente con el diagnóstico de disquinesia ciliar secundaria(16,17). De ellos, seis presentaron un número inusualmente elevado de cilias atípicas que mostraban alteraciones heterogéneas, tales como fusión ciliar, cilias múltiples, hernias de membrana (figura 4e) y ausencia de membrana plasmática con exposición del axonema (figura 4d). En los 11 pacientes restantes se observó una ausencia total de cilias y corpúsculos basales en las células epiteliales de los sectores de la vía respiratoria analizados (figura 4g). Estas alteraciones secundarias fueron especialmente frecuentes en las biopsias de cornete y en muchos casos impidieron descartar la existencia de una enfermedad ciliar primaria de base. En algunos casos esta situación se resolvió mediante el estudio de una biopsia bronquial, en la que generalmente se observó una menor incidencia de las alteraciones ciliares secundarias. Esto señala la relevancia diagnóstica de evaluar más de un sector de la vía respiratoria y, de ser posible, en más de una etapa de la vida del paciente(13).
Muchos autores consideran a las enfermedades ciliares secundarias como reversibles y, en tal sentido, estudios recientes realizados en cultivos celulares señalan que en condiciones favorables las células epiteliales de los pacientes portadores de alteraciones ciliares secundarias emiten cilias normales, que baten activamente(24-27). Se ha señalado que las alteraciones secundarias remiten con antibioticoterapia y el paso del tiempo(22), sin embargo, en la población analizada se observó la persistencia de alteraciones ciliares secundarias a tres o más semanas luego del último episodio infeccioso o alérgico, o ambos, y aun en respuesta al tratamiento con antibióticos. Esto indica que las lesiones ciliares inespecíficas persisten por períodos prolongados, por lo que su significado clínico no debe ser subestimado, ya que modifican localmente la eficiencia del transporte mucociliar, favoreciendo la recurrencia de la infección en las regiones afectadas.
A pesar de su sintomatología, siete de los pacientes estudiados presentaron una ultraestructura ciliar normal. Estos hallazgos son compatibles con la observación de que en ciertos casos las cilias con una estructura normal pueden presentar alteraciones funcionales que afectan la eficiencia del transporte mucociliar(2). Por esta razón, es necesario que los estudios de microscopía electrónica se complementen con estudios funcionales que permitan valorar la eficiencia del transporte mucociliar(28,29).
En su conjunto, los resultados obtenidos en la población estudiada apuntan a confirmar la alta frecuencia con que se presentan las afecciones ciliares en los pacientes respiratorios crónicos. Estos estudios señalan, además, que si bien el examen ultraestructural de las cilias no puede plantearse como un examen de rutina, debería estar indicado en los todos pacientes portadores de situs inversus y sus hermanos, así como en los pacientes portadores de afecciones respiratorias crónicas severas de inicio precoz carentes de diagnóstico etiológico y en los que se han descartado la fibrosis quística, los desórdenes inmunológicos y las deficiencias genéticas de ?1-antitripsina como causa primaria de la enfermedad respiratoria. Finalmente, nuestros estudios reafirman la relevancia de implementar métodos de diagnóstico complementarios a la microscopía electrónica que contribuyan a detectar afecciones funcionales de las cilias y permitan discriminar las alteraciones ciliares primarias, determinadas genéticamente, de las secundarias, provocadas por la injuria. Entre estos métodos cabe señalar, la valoración del movimiento ciliar in situ(28,29) e in vitro(27), los ensayos de ciliogénesis in vitro(24-26) y el recientemente desarrollado método de determinación de niveles de óxido nítrico(30,31). Es nuestro objetivo incorporar progresivamente estos nuevos métodos a nuestro laboratorio.
Agradecimientos
Al profesor Fernando Mañé Garzón y a la doctora Liria Martínez por mostrarnos el mundo de la patología ciliar. A todos los médicos e instituciones que confiaron en nuestro trabajo. Al doctor Federico Dajas por la revisión crítica de este manuscrito.
Summary
Primary ciliary dyskinesia is an inherited disease characterized by microanatomic or functional disorders affect to cilia or flagellum of the spermatozoa. Clinical signs include early recurrent infections of the aerial routes. Despite genetic determinants, recurrent infection and inflammation -as well as environmental agents- may lead to unspecific ciliar alterations and secondary ciliary dyskinesia.
Forty nasal and bronchial biopsies of 33 people suspected to carry ciliar dyskinesia were referred to the Cellular Biology Laboratory (Instituto de Investigaciones Biológicas Clemente Estable) and analyzed by electronic microscopy by transmission.
The overall study showed that 9 patients (27%) had abnormal ciliary ultrastructure compatible with primary ciliary dyskinesia diagnosis, of which three had situs inversus totalis. Secondary ciliary dyskinesia was seen in 52% of the patients and 21% of the patients showed, despite clinical signs, a normal ciliary ultrastructure. On average, results of the study tend to confirm high incidence of ciliar disorders in patients with chronic respiratory disorders. Considering that early diagnosis is essential in order to prevent permanent respiratory disorders, cilial ultrastructural examination should be considered in patients with situs inversus and sibling, as well as in patients with chronic and severe respiratory disease without ethyologic diagnosis.
Résumé
La dyskinésie ciliaire primaire est une maladie congénitale qui réunit un groupe hétérogène de conditions patholo-giques caractérisées par la présence de troubles micro-anatomiques ou fonctionnels, ou les deux, aux cils et aux flagèles spermatiques. Les manifestations cliniques de ce trouble sont variées et caractérisées par la présence précoce d'infection répétée des voies auditives-respiratoi-res. À part les déterminants génétiques, la propre infection et inflammation répétées -ainsi que quelques agents environnants- peuvent provoquer des troubles ciliaires. Ces troubles sont, pourtant, de type inespécifiques et mènent à ce qu'on appelle dyskinésie ciliaire secondaire. Dans ce travail on décrit les résultats obtenus de l'analyse de 40 biopsies de la mucose nasale et bronchiale provenant de 33 patients soupçonnés de porter une de ces formes de dyskinésie ciliaire, qui ont été remises par les médecins responsables au Laboratoire de Biologie cellulaire de l'Institut de Recherches Biologiques Clemente Estable pour son étude par microscopie électronique de transmission. Ces études ont montré que 9 patients (27%) présentaient des troubles ultrastructuraux ciliaires compatibles avec le diagnostic de dyskinésie ciliaire primaire. Trois parmi eux avaient situs inversus totalis. 52% des patients a présenté des troubles ciliaires secondaires. Malgré leurs manifesta-tions cliniques, 21% des patients restants a montré une ultrastructure ciliaire normale. Bref, les résultats obtenus dans la population étudiée visent à confirmer la haute fréquence de troubles ciliaires chez des patients respiratoi-res chroniques. Tenant compte que le diagnostic précoce de cette maladie est essentiel pour prévenir le développe-ment de lésions respiratoires irréversibles, on signale l'importance d'ordonner un examen ultrastructural des cils chez des patients porteurs de situs inversus et ses frères, ainsi que chez les individus atteints de cette maladie respiratoire chronique sévère à début précoce et n'ayant pas de diagnostic étiologique.
Bibliografía
1. Cowan MJ, Gladwin MT, Shelhamer JH. Disorders of ciliary motility. Am J Med Sci 2001; 321: 3-110.
2. Afzelius BA. Immotile cilia syndrome: past, present and prospect future. Thorax 1998; 53: 894-7.
3. Pedersen M, Rebbe H. Absence of arms in the axoneme of immobile human spermatozoa. Biol Reprod 1975; 12: 541-4.
4. Afzelius BA, Eliasson R, Johnsen O, Lindhomer C. Lack of dynein arms in immotile human spermatozoa. J Cell Biol 1975; 225-32.
5. Camner P, Mossberg B, Afzelius BA. Measurement of tracheobronquial clearance in patients with immotile cilia syndrome and its value in differential diagnosis. Eur J Resp Dis 1983; 64: 57-63.
6. Pedersen H, Mygind N. Absence of axonemal arms in nasal mucosa cilia in Kartagener's syndrome. Nature 1976; 262: 494-5.
7. Afzelius BA. A man syndrome caused by immotile cilia. Science 1976; 193: 317-9.
8. Eliasson R, Mossberg B, Camner P, Afzelius BA. The immotile cilia syndrome. A congenital ciliary abnormality as an etiologic factor in chronic airways infections and man sterility. N Engl J Med 1977; 297: 1-32.
9. Rossman Cm, Forrest JB, Lee RMKW, Newhouse MT. The dyskinetic cilia syndrome. Ciliary motility in immotile ciliary syndrome. Chest 1980; 78: 580-9.
10. Veerman AJP, Van der Baan A, Waltevreden EF, Leene W, Feenstra L. Cilia: immotile, dyskinetic, dysfunctional. Lancet 1980; 2(8188): 266-7.
11. Rott HD. Genetics of Kartagener's syndrome. Eur J Resp Dis 1983; 64 (suppl.127): 1-4.
12. Perraudeau M. Grand Rounds Hammersmith Hospital. Late presentation of Kartagener's syndrome. Br Med J 1994; 308: 519-21.
13. Holzman D, Ott PM, Félix H. Diagnostic approach to primary ciliary dyskinesia: a review. Eur J Pediatr 2000: 159: 95-8.
14. Bush A. Primary ciliary dyskinesia. Acta Otorhinolaryngol Belg 2000; 54: 317-24.
15. Teknos TN, Metson R, Chasse T, Malercia G, Dickersin GR. New developments in the diagnosis of Kartagener's syndrome. Otolaryngol Head Neck Surg 1997; 116: 68-74.
16. Bertransd B, Collet S, Eloy P, Rombaux P. Secondary ciliary dyskinesia in upper respiratory tract. Acta Otorhinolaryngol Belg 2000; 54: 309-16.
17. Mygind N, Pedersen M, Nielsen MH. Primary and secondary ciliary dyskinesia. Acta Otolaryngol 1983; 95: 688-94.
18. Rossman CM. Primary ciliary diskinesia: evaluation and management. Pediatr Pulmonol 1988; 5: 36-50.
19. Meeks M, Bush A. Primary ciliary dyskinesia. Pediatr Pulmonol 2000; 29: 307-16.
20. Date H, Yamashita M, Nagahiro I, Aoe M, Andou A, Shimizu N. Living-donor lobar lung transplantation for primary ciliary dyskinesia. Ann Thorac Surg 2001; 71: 2008-9.
21. Armengot M, Escribano A, Carda C, Sánchez C, Romero C, Basterra J. Nasal mucociliary transport and ciliary ultrastructure in cystic fibrosis. A comparative study with healthy volunteers. Int J Pediatr Otorhinolaryngol 1997; 40: 27-34.
22. Barlocco EG, Valleta EA, Canciani M, Lungarella G, Gardi C, De Santi MM, et al. Ultrastructural ciliary defects in children with recurrent infections of the lower respiratory tract. Pediatr Pulmonol 1991; 10: 11-7.
23. Meeks M, Bush A. Primary ciliary dyskinesia. Curr Pediatr 1998; 8: 231-6.
24. Gulisano M, Pacini S, Ruggiero M, Pacini A, Delrio AM, Pacini P. In vitro effects of some differentiation inductors in metaplasic epithelium of human nasal cavity. Cell Tissue Res 1996; 285: 119-25.
25. Jorissen M, Willems T, Van der Schueren B. Ciliary function analysis for diagnosis of primary ciliary dyskinesia: advantages of ciliogenesis in culture. Acta Otolaryngol 2000; 120: 291-5.
26. Jorissen M, Willems T, Van der Schueren B, Verbeken E. Secondary ciliary dyskinesia is absent after ciliogenesis in culture. Acta Otorhinolaryngol Belg 2000; 54: 333-42.
27. Jorissen M, Willems T, De Boeck K. Diagnostic evaluation of mucociliary transport: from symptoms to coordinated ciliary activity after ciliogenesis in culture. Am J Rhinol 2000; 14: 345-52.
28. Passali D, Bellussi L, Bianchini-Ciampoli M, De Seta E. Experiences in the dteremination of nasal mucociliary transport time. Acta Otolaryngol 1984; 97: 319-23.
29. Canciani M, Barlocco EG, Masetlla G, de Santi MM, gardi C, Lungarella G. The saccharin test meted for testing mucociliary function in patients suspected of having primary ciliary disquinesia. Pediatr Pulmonol 1988; 5: 210-4.
30. Karadag B, James AJ, Gültekin E, Wison NM, Bush A. Nasal and lower airway level of nitric oxide in children with primary ciliary dyskinesia. Eur Respir J 1999; 13: 1402-5.
31. Lefevre L, Willems T, Lidberg S, Jorissen M. Nasal nitric oxide. Acta Otorhinolaryngol Belg 2000; 54:271-80.

