










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Links relacionados
Compartilhar

 Permalink
PermalinkRevista Médica del Uruguay
versão On-line ISSN 1688-0390
Rev. Méd. Urug. vol.18 no.1 Montevideo maio 2002
Sr. Dr. Ariel Montalbán
Director de la Revista Médica del Uruguay
Esclerosis sistémica progresiva:
Dres. Rafael Pila Pérez1, Rafael Pila Peláez2,
Carmen Guerra Rodríguez3, Rita Díaz Leal3,
Margarita Pila Peláez4
Institución: Hospital "Manuel Ascunce Domenech". Camagüey. Cuba.
1. Especialista de II Grado en Medicina Interna. Profesor Titular.
2. Especialista de I Grado en Medicina Interna. Instructor.
3. Especialista de I Grado en Medicina General Integral. Policlínico Esmeralda.
4. Estomatólogo General. Policlínico de Previsora.
Correspondencia: Dr. Rafael Pila Pérez
General Gómez # 452
Camagüey. Cuba
C.P. 70100
La esclerosis sistémica progresiva (ESP) es un trastorno que afecta múltiples órganos y sistemas, y se caracteriza por grados diversos de cambios en vasos, fibrosis e inflamación de la piel y órganos internos. De manera característica la enfermedad pasa por varias fases de inflamación, fibrosis y atrofia(1). La duración de estas fases varía en diferentes órganos. La incidencia es tres veces mayor en el sexo femenino y la edad de aparición está entre los 25 y los 55 años, siendo muy rara su presentación antes de los 25 años y excepcional en la infancia(2).
El motivo de este manuscrito es la presentación de un caso de una niña de 8 años, con manifestaciones que ponen en duda si se trata o no de una afectación sistémica, lo cual, dada su edad, la colocaría entre los pocos casos descritos de ESP infantil.
Presentación del caso
Paciente de 8 años, mujer, desnutrida, disminución del panículo adiposo, talla 127 cm, peso 20 kg y en tercer percentil, refiere que desde los 7 años presentaba dolores articulares y pérdida de peso. Hace varios meses comenzó con dolores articulares que le impedían realizar labores, como escribir, y presentando fiebre de 38°C y 39°C, por lo que se ingresa para estudio.
Sin antecedentes personales ni familiares de interés.
Señala que desde hace ocho meses nota adormecimiento de ambas manos y pies, calambres nocturnos y dificultad para abrir y cerrar las manos así como dificultad para la marcha.
Examen físico
Fiebre de 38°C, palidez de piel y mucosas. La piel estaba endurecida, seca, lisa, adherida a planos profundos con manchas hipocrómicas en manos, flexura de los codos y rodillas. Zonas de retracción a nivel de los codos, rodillas y región cervical de aspecto escamoso. Las manos presentaban dedos afilados con ulceración a nivel del dedo medio de la mano derecha; dificultad para movilizar los miembros superiores e inferiores por ligera contractura en flexión (figura 1). Dificultad para abrir la boca, nariz afilada, pérdida de los pliegues faciales (facies de tapir). El pelo era escaso y quebradizo.
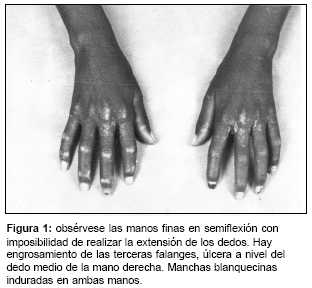
Aparato cardiorrespiratorio: no presentaba alteraciones. Hepatomegalia de 3 cm algo dolorosa y lisa. El resto del examen físico era normal.
Estudio analítico
Hemoglobina 8,5 g/L, leucocitos con fórmula normal, glicemia, creatinina, iones, enzimas, TASO, factor reumatoide, células LE(2), VDRL, proteína C reactiva, DNA, crioglobulinas, anticuerpos antimúsculo liso, complemento pruebas de Coombs directa e indirecta y conteo de reticulocitos: normales, velocidad de sedimentación 50 mm en primera hora. Constantes corpusculares: anemia ferropénica. Conteo de Addis: luecocituria. Urocultivo con antibiograma: Escherichia coli sensible a la ciprofloxacina. Radiografía de tórax: normal. Survey óseo: metarcarpianos lesiones, con tendencia a aspectos en "punta de lápiz", hiperdistensión de las muñecas y deflexión de los dedos y ambos codos. Ecografía abdominal: hepatomegalia de 3 cm, sin otras alteraciones. Radiografía de esófago. Estómago y duodeno: normal. Manometría esofágica no se pudo realizar. Radiografía de la boca: engrosamiento de la membrana periodontal. Electroforesis de proteínas: hipergamma globulinemia (1,98 g%). HLA: negativo. ECG, ecocardiograma y electromiografía: normales. Pruebas de la función hepática y respiratoria: normales. Biopsia de piel: la epidermis no muestra alteraciones y el dermis aparece aumentado de espesor y francamente colagenizado, con haces gruesos y empaquetados. No se advierten folículos pilosos ni infiltrado inflamatorio perceptible. Sólo se observa algunas glándulas sudoríparas comprimidas por el tejido conectivo colágeno (lesiones compatibles con esclerodermia). Con dieta, sales de hierro para la anemia, ciprofloxacina para la infección urinaria, así como sus manifestaciones dérmicas y articulares con el empleo de D-penicilamina a la dosis de 500 mg diarios, la paciente mejora. Durante tres años hemos seguido su evolución sin alteraciones, sólo con dolores articulares los cuales mejoran con el uso de colchicina.
La esclerodermia en bandas sin afectación sistémica es frecuente en el sexo femenino y puede comenzar en edades juveniles. La anatomía patológica pasa por dos estadios, pero en la mayoría de los cortes es frecuente que se entremezclen zonas en los dos estadios(3).
La piel se indura y adhiere más a las estructuras proximales, dificultando la motilidad, y en estas formas está afectado el músculo estriado que se atrofia y debilita de forma universal, dando signos en aparato locomotor, corazón, digestivo y sistema nervioso(1); nuestra paciente señalaba una afección del aparato locomotor.
La ESP tiene intensa afectación osteoarticular y de ella la tercera parte puede comenzar antes de tener lesiones de piel, con poliartralgias y poliartritis fundamentalmente a nivel interfalángico(4) como ocurrió en el caso que nos ocupa. En 90% de los enfermos, se presenta el fenómeno de Raynaud, esclerodactilia en 95%, calcinosis cutis en 89%, y 30% trastornos pigmentarios(1) como lo evidenció nuestra paciente.
El pronóstico de esta enfermedad es muy variable en su forma sistémica, pues hay formas graves en su supervivencia menor a los 10 años y hay otras de larga evolución que cursan con exacerbación a veces tardías.
El caso que nos ocupa nos deja en serias interrogantes pues no sabemos si catalogarlo como una ESP o como una esclerodermia localizada. Esta paciente presentó alteraciones osteomioarticulares y radiológicas del sistema óseo, todo ello precediendo a la aparición dérmica, lo cual es más específico de las formas sistémicas que de las localizadas(5).
Bibliografía
1. Sato S, Nagaoka T, Hasegawa M, Tamatani T, Naka Nishi T, Takigawa M, et al. Serum levels of connective tissue graonwth factor, elevated in patients with syspemic sclerosis: association with extent of skin sclerosis and severely of pulmonary fibrosis. J Rheumatol 2000; 27: 149-54.
2. Zakrzewska-Pniewska B, Jablonska S, Kowalska-Oledska E, Blaszczyk M, Hausmanowa-Petruise Wicz I. sympathetic skin respond in scleroderma, scleroderma overlap syndrome and inflammatory myophaties. Clin Rheumatol 1999; 18: 473-80.
3. Akagi A, Tajina S, Ishibashi A, Yamaguchi N, Nagai Y. Expression of type XVI collagen in human skin fibroblasts: enhanced expression in fibrotic skin disease. J Invest Dermatol 1999; 113: 246-50.
4. Poor Moghin H, Lucas M, Ferting N, Medsger T. Systemic Sclerosis sine sclerodermia: Demographic, clinical, and serologic features and survival in forty eight patients. Arthritis Rheum 2000, 43; 444-51.
5. Hanlow R, King S. Overview of the Radiology of connective tissue disorders in children. Eur J Radiol 2000; 33: 74-84.

