A propósito de un caso esporádico
Dres. Bernardo Hochmann*, Julio Coelho*, Ronald Salamano†
Cooperativa Médica de Rivera (COMERI), Rivera, Uruguay.
Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay
Resumen
Se presenta el caso clínico de una paciente de 66 años portadora de una encefalopatía espongiforme con criterios diagnósticos que configuran una enfermedad de Creutzfeldt-Jakob esporádica probable. Las abigarradas manifestaciones clínicas, junto a la baja incidencia de esta enfermedad, condujeron inicialmente a plantear diferentes diagnósticos diferenciales, hasta la aparición de demencia y mioclonias, junto con los complejos periódicos trifásicos en el electroencefalograma (EEG) y la presencia de proteína 14-3-3 en el líquido cefalorraquídeo (LCR) en la segunda muestra recolectada, que orientaron al diagnóstico definitivo.
Palabras clave: SÍNDROME DE CREUTZFELDT-JAKOB
Keywords: CREUTZFELDT-JAKOB SYNDROME
* Médico Neurólogo. Uruguay.
]]> † Profesor de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.Correspondencia: Dr. Bernardo Hochmann. 33 Orientales 1216. Rivera, Uruguay. Correo electrónico: hochmann@adinet.com.uy
Recibido: 28/10/11
Aprobado: 23/12/11
Introducción
Importancia del tema
Es esta una enfermedad rara en donde una proteína deletérea se replica sin participación del ácido desoxirribonucleico (ADN) o ácido ribonucleico (ARN), tiene un carácter infectante y puede atravesar la barrera de especies, estando comprobado que la ingesta de carne vacuna contaminada (encefalopatía bovina espongiforme) produce la variante de Creutzfeldt-Jakob (vCJ) en el hombre.
Si dicha circunstancia se concretara en nuestro país por encefalopatía bovina espongiforme autóctona, ocasionaría impensables consecuencias en la economía nacional.
Por todo ello, las enfermedades priónicas, a pesar de que sean raras, deben estar bajo la mira de neurólogos y autoridades de Salud Pública.
Las encefalopatías espongiformes transmisibles (EET), también llamadas enfermedades por priones debido al metabolismo aberrante de las proteínas priónicas, incluyen un grupo heterogéneo de enfermedades neurodegenerativas que afectan a humanos y animales.
]]> En los seres humanos existen formas esporádicas y familiares-genéticas de las EET, como la enfermedad de Creutzfeldt-Jakob (ECJ), la enfermedad de Gerstmann-Sträussler-Scheinker y el insomnio fatal familiar.También existen formas adquiridas como el kuru, formas iatrogénicas y la vCJ.
En los animales se observan el scrapie en ovinos, la encefalopatía espongiforme en bovinos, la encefalopatía del visón y la encefalopatía del alce(1).
La etiopatogenia de las EET se debería a un cambio en la conformación de una proteína normal celular (Pr pc) de significado aún no aclarado en la fisiología celular, que en contacto con la misma proteína, que tiene un plegamiento anómalo (Pr EET), generaría un proceso acumulativo de esta proteína que resulta ser deletéreo para el sistema nervioso central. Prusiner denominó a esta proteína anómala como PRION (PRoteinaceus InfectiOus ageNt). Existen factores favorecedores para este cambio conformacional y ello puede permitir que dentro del Creutzfeldt-Jakob esporádico y otras enfermedades priónicas en el ser humano existan diferentes expresiones fenotípicas clínicas, electroencefalográficas y de la proteína 14-3-3 en el LCR (elementos claves para el diagnóstico). La más importante sería el polimorfismo en el codón 129 del brazo corto del cromosoma 20, que codifica metionina (M) o valina (V). Así se ha visto que 70% de los Creutzfeldt-Jakob esporádicos son metionina-metionina (MM) en el codón 129 y que en el Creutzfeldt-Jakob iatrogénico el 90% son homocigotos en el codón 129 (MM o valina-valina, VV)(2).
La ECJ es una encefalopatía subaguda y progresiva, poco frecuente, con una incidencia de un caso por millón de habitantes por año(1). Se conocen formas esporádicas de la enfermedad (que reúne a 85%-90% de los casos) y formas familiares-genéticas (10%-15% restantes).
La incidencia de la ECJ en Uruguay fue de 0,7 casos / 1.000.000 habitantes / año para el período comprendido entre 1997 y 2009, considerando las formas probables y definitivas.
La ECJ esporádica en nuestro país fue la forma más frecuente, 81% de los casos, mientras que la forma hereditaria fue de 19%. La edad de aparición para las formas esporádicas fue de una media de 61,6 años.
De las formas hereditarias se detectaron dos familias, la primera con la mutación G114V y la segunda con la mutación E200K(3,4). La principal diferencia entre ambas mutaciones fue la edad de aparición, siendo la primera de aparición precoz (segunda y tercera década de la vida) y la segunda en pacientes adultos-adultos añosos.
En Chile se han observado familias con mutaciones dobles del codón 200 y 129, que suponen un acercamiento a la explicación de la mayor incidencia de esta enfermedad (dos casos por millón de habitantes por año), mientras que en Brasil se ha observado uno de los siete casos mundiales de ECJ familiar con la mutación en el codón 210(5,6).
Las formas iatrogénicas y de la vCJ no han sido detectadas en Uruguay(3).
]]> La presentación clínica clásica incluye deterioro cognitivo a forma demencial rápidamente progresiva, mioclonias y ataxia. La evolución es siempre mortal en un período, en general, menor a dos años. Al inicio de la enfermedad pueden presentarse trastornos visuales y elementos cerebelosos.La paraclínica que ayuda al diagnóstico incluye el EEG con complejos periódicos de ondas agudas, por lo general trifásicas, y el estudio de proteína 14-3-3 en el LCR, que suele ser positivo aunque no específico de la enfermedad (1,2).
Los criterios diagnósticos para la ECJ recomendados por la Organización Mundial de la Salud (OMS) son:
1. Con características neuropatológicas o confirmación de la proteína priónica (PRNP).
1. Con demencia progresiva + dos de (mioclonias, problemas visuales/cerebelosos, trastornos piramidales/extrapiramidales, mutismo aquinético) + EEG típico + duración < a 2 años.
2. Con demencia progresiva + dos de (mioclonias, problemas visuales/cerebelosos, trastornos piramidales/extrapiramidales, mutismo aquinético) + duración < 2 años + proteína 14-3-3 positiva.
1. Con demencia progresiva + dos de (mioclonias, problemas visuales/cerebelosos, trastornos piramidales/extrapiramidales, mutismo aquinético) + duración < 2 años(2).
Objetivo
El objetivo principal del presente trabajo es publicar el primer caso de ECJ en nuestro país con sus características clínicas y paraclínicas.
Caso clínico
Presentamos el caso clínico de una ECJ esporádica, estableciendo las dificultades diagnósticas que aparecen en su primera etapa de desarrollo.
NP, paciente de sexo femenino, de 66 años, con antecedentes personales de hipertensión arterial (HTA) que inició su sintomatología en octubre de 2010 con una evolución progresiva caracterizada por cefaleas holocraneanas, disminución de la agudeza visual, diplopía horizontal y trastorno del equilibrio.
Al examen físico se encontró un síndrome cerebeloso vermiano y hemisférico izquierdo caracterizados por un aumento de la base de sustentación, asinergia y adiadococinesia.
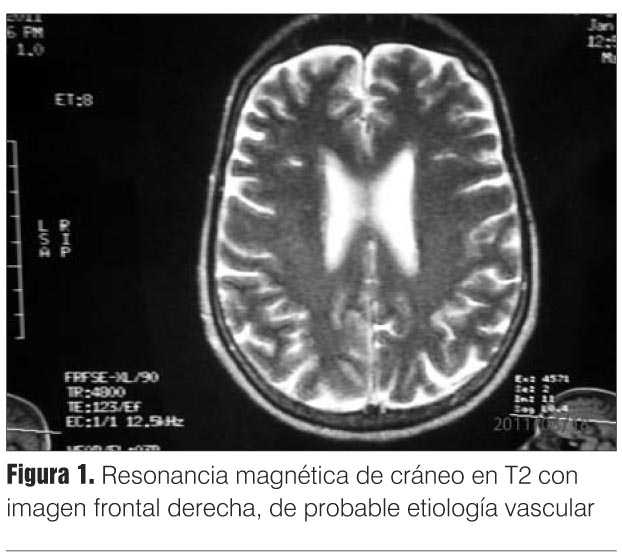
Con el planteo de proceso expansivo infratentorial lateralizado a izquierda, se solicitó resonancia magnética (RM) de cráneo que descartó esta hipótesis diagnóstica mostrando un aumento de la señal en Flair y T2 en región frontal derecha de probable etiología vascular (figura 1).
]]> La paciente agregó rápidamente mioclonias bilaterales, incontinencia urinaria y se constató un síndrome motor piramidal de liberación, bilateral, con hiperreflexia y clonus.Se solicitaron estudios inmunoquímicos del LCR (proteína básica de la mielina, distribución oligoclonal y presencia de cadenas livianas) y anticuerpos onconeuronales.
Una nueva RM de cráneo y cervical no aportaron para el diagnóstico. No pudo realizarse RM con difusión. También se solicitaron en plasma, anticuerpos anti ADN, anti SSa, anti SSb, anti SM y TSH, Ac antinucleares, PEF, Ac antihepatitis B y C, Ac antifosfolipídicos, dosificación vitamina B12 y ácido fólico.
El estudio inmunoquímico del LCR mostró un marcado aumento del índice de necrobiosis neuronal y de actividad inmunoalérgica con inmunocomplejos solubles (por exceso de antígeno).
Se solicitaron estudios de búsqueda de neoplasma oculto con tomografía computada (TC) de abdomen, tórax, ecografía de abdomen y pélvica, todos los cuales fueron normales.
La paciente tuvo una mala evolución clínica con episodios de confusión, alucinaciones visuales, tendencia al sueño, acentuación de las mioclonias tanto espontáneas como provocadas, agregando posteriormente disfagia e intenso síndrome extrapiramidal de tipo tónico frontal bilateral, llegando al mutismo aquinético y tetraplejia.
Ante la variación del cuadro clínico y el empeoramiento del mismo, que hicieron presumir la existencia de una enfermedad priónica o eventualmente una encefalitis viral de curso crónico, se solicitaron EEG, estudio de la proteína 14-3-3 y relevamiento viral en LCR.
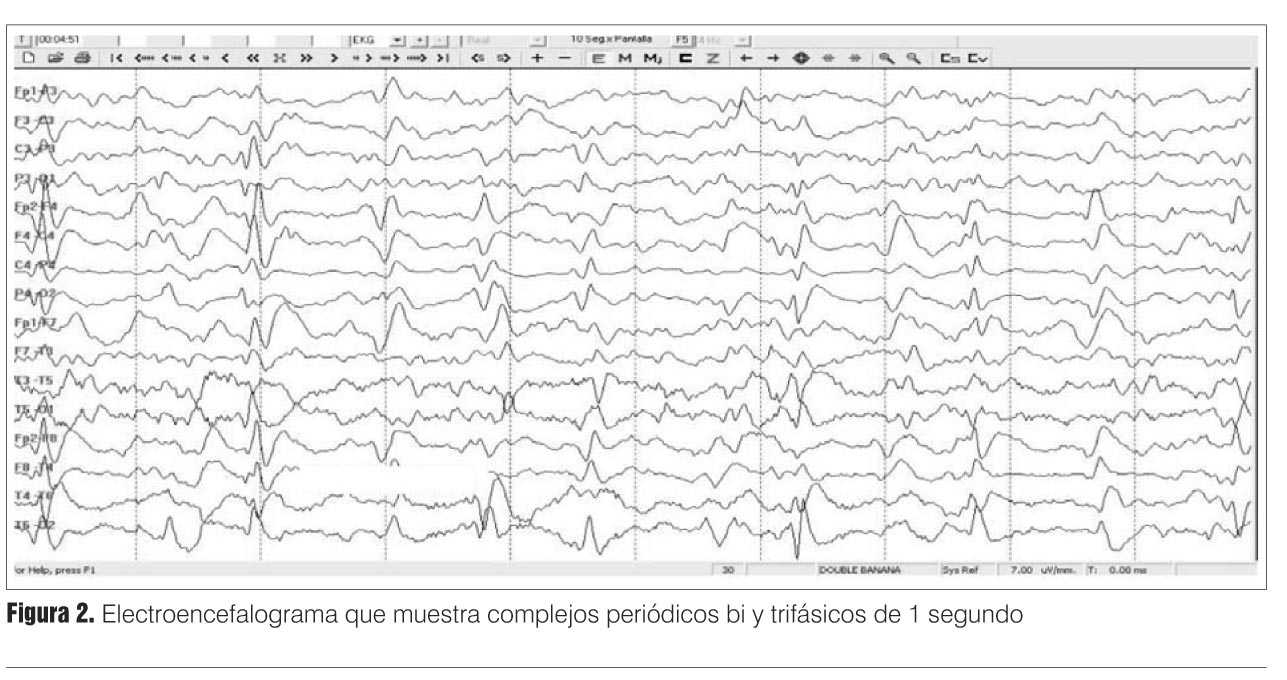
El EEG mostró un trazado con complejos semiperiódicos de punta onda lenta, de 1-2 ciclos/seg, en algunas oportunidades trifásicos, bilaterales, con ligero predominio a derecha (figura 2). Este trazado orientó el cuadro hacia una encefalopatía subaguda como las priónicas.
El estudio de la determinación de la proteína 14-3-3 fue negativo en primera instancia y positivo en segunda instancia (nueve días después). El relevamiento viral en LCR para familia herpes y enterovirus fue negativo.
Se planteó el diagnóstico de ECJ probable teniendo en cuenta su deterioro cognitivo rápidamente progresivo, la presencia de mioclonias, síndromes cerebeloso, extrapiramidal y piramidal con mutismo aquinético, más un EEG típico y la presencia de proteína 14-3-3 en LCR.
]]> La evolución del cuadro clínico se mantuvo con su tendencia al deterioro de vigilia, con fluctuaciones que variaban según situación metabólica-infecciosa. Mantuvo su tetraplejia con hipertonía extrapiramidal. Presentó varios episodios de infecciones del aparato urinario y respiratorio.La paciente falleció luego de nueve meses de evolución. No existen casos similares en su familia.
El tratamiento ensayado desde el comienzo del cuadro, pasando por las distintas etapas evolutivas del mismo, incluyó la administración de metilprednisolona, amitriptilina, ketoprofeno, aciclovir, alimentación parenteral, antibióticos, antiepilépticos, clonazepan y heparina de bajo peso molecular.
Discusión
Las vicisitudes clínicas experimentadas en este caso suelen ocurrir en este tipo de enfermedades, ya sea por su bajísima frecuencia como por lo abigarrado de su cuadro clínico.
En este sentido debemos mencionar que el inicio de este caso, con síntomas referidos a la fosa posterior como diplopía, disminución de la agudeza visual, cefaleas y ataxia, se corresponde con una combinación de las formas atáxica y de Heidenheim de la ECJ(1).
Son entendibles las dificultades diagnósticas iniciales dado el inicio de la signo-sintomatología en fosa posterior, pero el viraje a la agravación rápida con deterioro de conciencia, agregando en semanas mioclonias, mutismo aquinético y tetraplejia, hicieron pensar en una enfermedad priónica y sus diagnósticos diferenciales.
A esta altura, los estudios paraclínicos solicitados, en búsqueda de la confirmación etiológica de lo que ya se constataba como una encefalopatía subaguda, demostraron el abanico de posibilidades que se abren al médico enfrentado a este tipo de enfermedades.
De hecho, al comienzo del relevamiento etiológico se planteó, a través del estudio inmunoquímico del LCR, la posibilidad de encontrarnos frente a una encefalopatía paraneoplásica. Los otros diagnósticos diferenciales señalados en la literatura abarcan diferentes tipos de demencia (Alzheimer, frontotemporal, por cuerpos de Lewy, vascular) así como encefalopatías tóxicas (litio, bismuto), encefalitis virales, etcétera, que fueron descartadas por la historia clínica o los exámenes realizados.
Un detalle interesante es el valor jerárquico del EEG en la evolución de este tipo de encefalopatías. El EEG suele ser muy inespecífico en etapas tempranas de la ECJ.
]]> Ya en etapas medianas y tardías suelen observarse los complejos periódicos trifásicos de un ciclo/segundo. Estos complejos periódicos se observan en 2/3 de los pacientes con ECJ esporádica, teniendo un valor predictivo de 95%(2).Están presentes, en general, en pacientes homocigotas en el codón 129 para metionina MM o heterocigotas MV y tienen una sensibilidad de 80% para el fenotipo MM1 y de 75% para el fenotipo MV1(2,7).
El EEG típico de Creutzfeldt-Jakob es considerado un criterio diagnóstico útil para reclasificar un caso de posible a probable.
La falta de especificidad del patrón típico del EEG se debe a que puede encontrarse en otras patologías que plantean el diagnóstico diferencial con la ECJ en algún momento de su evolución, como la enfermedad de Alzheimer, demencia por cuerpos de Lewy, Bingswanger, MELAS, encefalopatía posanóxica, encefalopatía hepática, hipo o hipernatremia, etcétera(2).
Lo mismo ocurre con el estudio de la proteína 14-3-3 en el LCR donde, como en este caso, fue negativa al inicio del cuadro y positiva a los nueve días de la primera toma.
La proteína 14-3-3 es una proteína intracitoplasmática que se usa como marcador de daño neuronal subagudo, útil en el contexto clínico de la ECJ.
Hay evidencia que sugiere que el marcador 14-3-3 positivo en el LCR es más factible de encontrar en las etapas intermedias de la enfermedad, sin embargo esto no debería condicionar el momento de punción lumbar, ya que la detección de 14-3-3 puede obtenerse en todas las fases de la enfermedad y estaría relacionada con el subtipo de Creutzfeldt-Jakob esporádico(2).
La proteína 14-3-3 puede detectarse en procesos agudos como infartos o encefalitis (especialmente herpética), pero no en demencias de curso subagudo o crónico, por lo que su valor diagnóstico a favor de una ECJ es muy alto(8).
En el contexto clínico de Creutzfeldt-Jakob, la detección de proteína 14-3-3 en LCR tiene una alta sensibilidad y especificidad (entre 90% a 97% y entre 87% a 100%, respectivamente)(2,9).
La OMS y el programa europeo para la vigilancia de Creutzfeldt-Jakob han incluido el hallazgo de proteína 14-3-3 positivo en LCR por inmunoblot o Elisa, entre los criterios diagnósticos de Creutzfeldt-Jakob.
]]> En pacientes que tienen demencia progresiva de menos de dos años y otros criterios de Creutzfeldt-Jakob posible, un LCR positivo para la proteína 14-3-3 tiene un peso diagnóstico igual al del EEG específico, permitiendo reclasificar un caso posible como probable(2).En cuanto a la RM, esta no es un criterio de la OMS para el diagnóstico de ECJ esporádica probable hasta el momento actual, a diferencia del EEG y de la proteína 14-3-3.
Se considera una RM típica de ECJ esporádica si existe incremento de señal en caudado y el putamen asociada o precedida de hiperintensidad cortical. Las secuencias de difusión son las más apropiadas, seguidas por Flair. Cuando se detecta hiperintensidad en el pulvinar, esta debe ser siempre menor que la que se observa en los núcleos de la base(2).
La v ECJ difiere de la ECJ esporádica por la media de edad de comienzo que es de 27 años para la primera y de 60 años para la segunda.
A su vez, la duración de la enfermedad es más prolongada para la v ECJ (media de 14 meses), que para la ECJ esporádica (media de 8 meses).
En cuanto a la clínica podemos decir que la v ECJ se presenta con trastornos psiquiátricos y dolores francos o disestesias. Luego agrega síntomas motores y, por último, demencia.
En la patogenia de la infección por vía oral que ocasiona la v ECJ, el agente infectante se diseminaría desde el tejido linfático del intestino hacia otros tejidos linfoides, incluso el bazo. Del bazo hacía la médula espinal y al cerebro, vía retrógrada a través de las fibras nerviosas de los nervios esplácnicos.
El agente también puede diseminarse en forma retrógrada del plexo parasimpático a la pared intestinal por el vago y penetrar en el tronco cerebral por el nervio motor del vago(2).
Dada la posibilidad de transmisión de esta enfermedad por instrumentos neuroquirúrgicos, la OMS recomienda no efectuar una biopsia, excepto cuando se plantea el diagnóstico diferencial con casos tratables (por ejemplo, encefalitis viral). El diagnóstico definitivo, que es anátomo-patológico, necesita de un laboratorio de alta seguridad para poder realizarlo, mecanismo con el que no contamos en nuestro país. Según diferentes estudios, el diagnóstico de probable se acerca en 90% al definitivo(2).
Summary
]]> The study presents the clinical case of a 66 year old patient carrier of a spongiform encephalopathy with diagnostic criteria matching a probable sporadic Creutzfeldt-Jakob disease.These variegated clinical manifestations, together with the low incidence of the disease led us to initially consider several differential diagnoses. Yet, the arousal of dementia and myoclonus and triphasic periodic complexes evidenced in the electroencephalogram and the presence of 14-3-3 protein in the
Cephalorachidian fluid in the second sample collected contributed to the final diagnosis.
Resumo
Apresenta-se o caso clínico de uma paciente de 66 anos portadora de uma encefalopatia espongiforme com critérios diagnósticos de uma provável doença de Creutzfeldt-Jakob esporádica. As manifestações clínicas heterogêneas e a baixa incidência desta patologia levaram inicialmente a considerar diferentes diagnósticos diferenciais. O surgimento de sintomas de demência e mioclonias, associados aos complexos periódicos trifásicos no eletroencefalograma (EEG) e a presença de proteína 14-3-3 no líquido cefalorraquideano (LCR) na segunda amostra obtida orientaram ao diagnóstico definitivo.
Bibliografía
1. Zivkovic S, Boada M, López O. Revisión de la enfermedad de Creutzfeldt-Jakob y otras enfermedades priónicas. Rev Neurol 2000; 31(12): 1171-9.
2. Begué C, Piccardo P, Taratuto A. Encefalopatías espongiformes transmisibles: enfermedad de Creutzfeldt-Jakob. In: Salamano R, Scavone C, Wajskopf S, Savio E. Neuroinfecciones en el adulto y el niño. Montevideo: Editorial Arena, 2008. p. 203-11.
]]>3. Lewin S, Perna A, Salinas D, Ketzoian C, Vernengo L, Rodríguez MM, et al. Enfermedades priónicas en el ser humano en Uruguay: registro de los últimos 25 años. In: IV Congreso Uruguayo de Neurología, Neuropunta 2010. Punta del Este, 29 nov. - 3 dic. 2010. Montevideo: Sociedad Uruguaya de Neurología, 2010.
4. Rodríguez MM, Peoc'h K, Haïk S, Bouchet C, Vernengo L, Mañana G, et al. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology 2005; 64(8): 1455-7.
5. Cartier L, Fernández J, Ramírez E. Forma familiar de la enfermedad de Creutzfeldt-Jakob: marcadores genéticos en 4 familias chilenas. Rev Méd Chile 2006; 134(9): 1116-22.
6. Huang N, Marie S, Kok F, Nitrini R. Familial Creutzfeldt-Jakob disease associated with a point mutation at codon 210 of the prion protein gene. Arq Neuropsiquiatr 2001; 59(4): 932-5.
7. Zerr I. Clinical manifestations of prion diseases in humans. Arch Inst Neurol 2001; 4-1: 41-3.
]]>8. Piccardo P. Enfermedades priónicas: en qué estamos hoy? Neurodegeneración. Disponible en: http://www.institutodeneurologia.edu.uy/sitio/documentos/picardo.pdf [Consulta: 10/06/11]
9. Zerr I. Clinical diagnostic test in human prion disease. Arch Inst Neurol 2001; 4-1: 45-7.
]]>
{kind=link}
{kind=link}