CASO CLÍNICO
Enfermedad de Krabbe.
A propósito de un caso clínico
Dres. Ricardo Tambasco1, Loreley García2, Alfredo Cerisola3, Gustavo Giachetto4, Virginia Kanopa5, Aída Lemes6
1. Residente de Pediatría. Departamento de Pediatría y Especialidades. Facultad de Medicina. UdelaR.
2. Profesora Agregada de Pediatría. Departamento de Pediatría y Especialidades. Facultad de Medicina. UdelaR.
3. Profesor Agregado de Neuropediatría. Cátedra de Neuropediatría. Facultad de Medicina. UdelaR. ]]>
4. Profesor de Clínica Pediátrica. Departamento de Pediatría y Especialidades. Facultad de Medicina. UdelaR. Responsable del Programa Nacional de Salud de la Niñez. Departamento de Planificación Estratégica en Salud, MSP.
5. Profesora Agregada de Pediatría. Departamento de Pediatría y Especialidades. Facultad de Medicina. UdelaR. Neuropediatra.
6. Médica. Instituto de Genética Médica. Hospital Italiano, Montevideo.
Fecha recibido: 5 de junio 2012-
Fecha aprobado: 26 de julio 2012
Resumen
La enfermedad de Krabbe es una afección genética con mecanismo de herencia autosómico y recesivo de muy baja incidencia. Está determinada por deficiencia en la actividad de la enzima lisosomal galactocerebrósido-b-galactosidasa (o galactosilcerebrosidasa). Tiene diferentes formas clínicas: infantil, juvenil y de la edad adulta. En Uruguay las enfermedades genéticas son una causa importante de morbi-mortalidad. El reconocimiento por parte del pediatra de elementos clínicos en forma precoz, es clave en el diagnóstico oportuno. Se presenta el caso de una niña con la forma infantil de enfermedad de Krabbe cuyas manifestaciones clínicas se iniciaron a los 7 meses de vida y el diagnóstico se confirmó a los 10 meses por dosificación de la actividad galactocerebrósido-b-galactosidasa. El objetivo de esta comunicación es jerarquizar el reconocimiento oportuno de las manifestaciones clínicas, revisar los conocimientos actuales en relación al diagnóstico, incluyendo el diagnóstico prenatal, en vistas al asesoramiento genético.
Palabras clave:
LEUCODISTROFIA DE CÉLULAS ]]>
GLOBOIDES
ESFINGOLIPIDOSIS
ERRORES INNATOS DEL METABOLISMO
Summary
Krabbe disease is a genetic condition with recessive autosomal inheritance mechanism and very low incidence. Is determined by deficiency in activity of the lysosomal enzyme beta-galactosidase-galactocerebroside (or galactosilcerebrosidasa). Has different forms: children, youth and adulthood. In Uruguay genetic diseases are a major cause of morbidity and mortality. The recognition by the pediatrician in an early clinical elements, is key to early diagnosis. A case of a girl whose clinical manifestations began at 7 months and the diagnosis was confirmed at 10 months by activity assay of galactocerebroside-beta-galactosidase. The aim of this paper is to prioritize the timely recognition of the clinical manifestations, review current Knowledge regarding the diagnosis, including prenatal diagnosis, genetic counseling in view.
Introducción
La enfermedad de Krabbe (EK) es una afección genética con mecanismo de herencia autosómico y recesivo descrita por primera vez en 1916. En ese año Krabbe describe los elementos clínicos e histológicos en dos hermanos que habían fallecido de lo que él definió como una esclerosis cerebral difusa familiar infantil (1). Él describe la encefalopatía rápidamente progresiva con muerte temprana y espasticidad y señala la ocurrencia familiar. Además realiza un detallado análisis de las células globoides, el marcador histológico de la enfermedad. En 1970 -1971, se describe la deficiencia enzimático básica de la EK: galactosilcerebrosidasa o galactocerebrósido-b-galactosidasa (2,3). Se trata de deficiencia en la actividad de una enzima que normalmente se ubica y ejerce su función catabólica en el interior de los lisosomas.
Dentro del capítulo de las enfermedades por deficiencia de actividad de una enzima lisosomal, la EK pertenece al grupo de la esfingolipidosis junto a otras afecciones por errores congénitos del metabolismo como: Gaucher, Tay-Sachs, Niemann-Pick A y B entre otras.
La EK ha sido reportada universalmente. Se ha estimado una incidencia de 1 cada 170.000 nacidos vivos. La máxima incidencia ha sido comunicada en la comunidad Druze israelí (6 por 1.000 nacidos vivos) (4).
]]> La edad de comienzo determina diferentes formas clínicas: infantil, juvenil y de la edad adulta. La forma infantil es la más frecuente, la cual clínicamente se manifiesta en general en forma temprana, durante los primeros meses de vida, en niños aparentemente sanos. Suele presentarse antes de los 6 meses, con llanto frecuente, irritabilidad marcada, dificultad en la alimentación, vómitos y regresión a nivel del desarrollo psicomotor (5-7). La deficiencia enzimática, afecta específicamente la sustancia blanca ya que interviene en el catabolismo lisosomal de macromoléculas complejas como son ciertos esfingolípidos, principalmente del galactosilcerebrósido, pero también de galactosilesfingosina (o psicosina). El déficit conduce a la acumulación del metabolito citotóxico (psicosina) que altera el funcionamiento de las mitocondrias y peroxisomas generando la apoptosis de las células formadoras de mielina como oligodendrocitos. La consecuencia es una desmielinización que afecta en forma primaria al sistema nervioso central y posteriormente también al sistema nervioso periférico (8). Esta patología consiste más en una destrucción de la mielina que en una formación anormal de la misma (9).Por el momento no se cuenta con un tratamiento específico, el mismo es sintomático.
Su carácter progresivo e irreversible exige un reconocimiento tempranoy diagnóstico oportuno, en vistas al asesoramiento genético dado el alto riesgo de recurrencia en futura descendencia de la pareja.
Uruguay, al igual que el resto de los países en vías de desarrollo, experimenta un descenso sostenido en la tasa de la mortalidad infantil y un cambio epidemiológico, constituyendo las enfermedades genéticas una causa importante de morbimortalidad. En los últimos 2 años estas enfermedades representan la segunda causa de mortalidad infantil, tanto en el período neonatal como posneonatal (10). La EK, aunque poco frecuente, constituye una de las enfermedades neurodegenerativas relacionadas con errores congénitos del metabolismo que es importante conocer.
Se presenta el caso de una niña cuyas manifestaciones clínicas se iniciaron a los 7 meses de vida y el diagnóstico se confirmó a los 10 meses por dosificación de la actividad galactocerebrósido-b-galactosidasa. El objetivo de esta comunicación es jerarquizar el reconocimiento oportuno de las manifestaciones clínicas, revisar los conocimientos actuales en relación al diagnóstico, incluyendo el diagnóstico prenatal, en vistas al asesoramiento genético.
Caso clínico
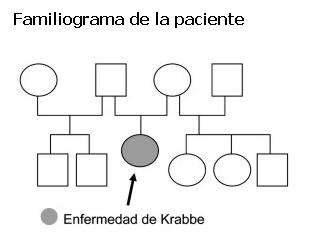
Paciente de 10 meses, sexo femenino, procedente de Rocha, primera hija de la pareja de padres no consanguíneos. Padre de 62 años de edad y madre de 28 al momento del nacimiento de la paciente. No se aprecia situación similar en la familia. En la figura 1 se muestra el familiograma. Embarazo bien controlado, parto a término, eutócico. Peso al nacer: 3.230 g, perímetro cefálico (PC): 33 cm, talla: 50 cm. Sin patología perinatal. Controlada en salud, esquema de vacunación vigente, con buen crecimiento y desarrollo hasta los 7 meses. Posteriormente, pérdida de conductas adquiridas sin nuevos logros en las diferentes áreas del desarrollo. A los 10 meses presentaba pobre seguimiento visual, sin sonrisa social, hipotonía generalizada que dificulta la sedestación y el sostén cefálico. No balbuceo ni laleo. No toma objetos. Irritabilidad excesiva. Desde los 7 meses no había habido ganancia de peso, presenta vómitos reiterados y dificultad en la alimentación.
 ]]>
Ingresa al Hospital Pediátrico del Centro Hospitalario Pereira Rossell (CHPR), derivada por pediatra tratante. Al examen se destaca: paciente reactiva. Facies inexpresiva. Irritable, llanto monótono. Peso: 7,350 g (P3-P15), talla: 66 cm (P3-P15), PC: 44 cm (P15-P50). Piel y mucosas: sin lesiones en piel, normocoloreadas. Cardiovascular: ritmo regular 120 cpm, ruidos bien golpeados. No soplos. Pleuropulmonar: FR 50 rpm. No tirajes. Buena entrada de aire bilateral. Abdomen: blando, depresible, sin visceromegalias. Psiconeuromuscular: Glasgow 15. Pupilas simétricas y reactivas. No fija ni sigue con la mirada. Hipotonía axial de los músculos flexores, hipertonía de los cuatro miembros a forma de cuadriparesia espástica con pulgares incluidos, reflejos vivos, Babinski bilateral. Fontanela anterior normotensa con latido. Resto del examen físico sin alteraciones.
]]>
Ingresa al Hospital Pediátrico del Centro Hospitalario Pereira Rossell (CHPR), derivada por pediatra tratante. Al examen se destaca: paciente reactiva. Facies inexpresiva. Irritable, llanto monótono. Peso: 7,350 g (P3-P15), talla: 66 cm (P3-P15), PC: 44 cm (P15-P50). Piel y mucosas: sin lesiones en piel, normocoloreadas. Cardiovascular: ritmo regular 120 cpm, ruidos bien golpeados. No soplos. Pleuropulmonar: FR 50 rpm. No tirajes. Buena entrada de aire bilateral. Abdomen: blando, depresible, sin visceromegalias. Psiconeuromuscular: Glasgow 15. Pupilas simétricas y reactivas. No fija ni sigue con la mirada. Hipotonía axial de los músculos flexores, hipertonía de los cuatro miembros a forma de cuadriparesia espástica con pulgares incluidos, reflejos vivos, Babinski bilateral. Fontanela anterior normotensa con latido. Resto del examen físico sin alteraciones.
De la paraclínica solicitada se destaca:
Gasometría, ionograma con cloremia, glicemia y amonio: normales. Pesquisa neonatal ampliada por espectrofotometría de masas en tándem en gotas de sangre en papel de filtro: perfil de aminoácidos y perfil de acilcarnitinas normales.
El estudio citoquímico del líquido cefalorraquídeo (LCR) mostró: aspecto límpido sin coágulos, sedimento nulo, proteínas: 0,78 g/l, Glucosa: 0,53 g/l, Lactato 1,88 mMol/l (0,6-2,8). 0 elementos.
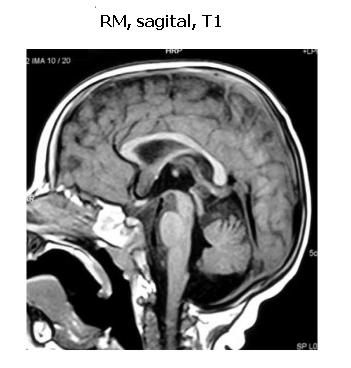
En la resonancia magnética (RM) de cráneo: parénquima encefálico de morfología conservada con adecuada distribución de la sustancia blanca y gris. En la sustancia blanca de centros ovales se identifica alteración en la intensidad de la señal, caracterizada por hipointensidad en T1 e hiperintensidad en las secuencias ponderadas en T2. Esta alteración es bilateral y simétrica, sin efecto de masa, compatible con desmielinización (figuras 2 y 3).

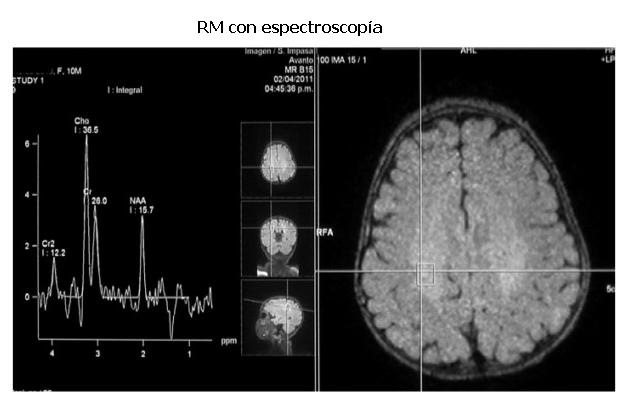
RM con espectroscopía: alteración en los picos obtenidos, caracterizados por disminución de la N-acetil-aspartato, con aumento de la colina y de los picos de lactato (figura 4).
Estudio electrofisiológico: alteración de las velocidades de neuroconducción periférica con participación mielínica y axonal.
En el estudio dinámico de la deglución se confirmó alteración del tercer tiempo con pasaje de escasa cantidad de contraste hacia vías respiratorias altas y bajas.
Con el diagnóstico clínico de encefalopatía progresiva de inicio en el primer año de vida, caracterizado por regresión psicomotriz, irritabilidad, cuadriparesia espástica, alteración sensorial visual, sumado a los hallazgos neuroimagenológicos de compromiso de sustancia blanca periventricular y profunda, asociada a hiperproteinorraquia, alteraciones de las vías visuales y auditivas, y de las velocidades de conducción nerviosa periféricas, se planteó, en primer lugar, que pudiera corresponder a una EK o a una leucodistrofia metacromática, y se solicitó dosificación de la actividad galactocerebrósido-b-galactosidasa y Aril-sulfatasa A en leucocitos y dosificación de sulfátidos en orina. Los resultados fueron: galactocerebrósido-b-galactosidasa: Valor hallado: 0,80 nmol/h/mg de proteínas (valor de referencia: 11,0–39,0 nmol/h/mg de proteínas). Arilsulfatasa «A»: Valor hallado: 8,40 nmol/h/mg de proteínas (valor de referencia: 7,40 – 17,30 nmol/h/mg de proteínas). La excreción de sulfátidos en orina mostró valores dentro de los rangos normales.
Discusión
]]> Se trata de una lactante de 10 meses, morfológicamente normal, con pérdida de los hitos del desarrollo, detención del crecimiento y cambio de conducta habitual dado por irritabilidad en los últimos meses. Se destaca del examen físico un síndrome piramidal a forma de cuadriparesia espástica, estos hallazgos orientan a una enfermedad neurológica progresiva de origen metabólico, un error congénito del metabolismo. Frente a este planteo es necesario recurrir al estudio imagenológico para orientar al tipo de enfermedad.Distintos autores sostienen que la RM de cráneo en oportunidades resulta el estudio de elección frente al planteo etiológico de un error congénito del metabolismo tal como se evidenció en este caso. Su resultado fue muy orientador de una enfermedad desmielinizante, al ser analizado junto a la historia clínica, elementos del examen físico, disminución de velocidad de conducción e hiperproteinirraquia. Con estos elementos, se plantea diagnóstico de probable leucodistrofia. Entre ellas, en primer lugar enfermedad de Krabbe y en segundo lugar leucodistrofia metacromática. En este caso se confirmó el diagnóstico de EK a través de la dosificación de la actividad galactocerebrósido-b-galactosidasa en leucocitos.
La enfermedad de Krabbe es producida por el déficit congénito de la enzima galactocerebrósido-b-galactosidasa que origina la acumulación de sus dos sustratos: galactocerebrósido y psicosina. Esta deficiencia altera el metabolismo de la mielina, ocasionando el depósito intracelular progresivo de galactocerebrósido a nivel de los macrófagos, principalmente en el sistema nervioso central, formando las células globoides patognomónicas de esta enfermedad, ocasionando la muerte celular y el consiguiente daño tisular (6).
Desde la perspectiva patogénica, el aumento de la psicosina produce efecto tóxico que lleva a la muerte celular temprana por medio de la alteración del metabolismo de las mitocondrias, de los peroxisomas y la inducción de la apostosis. El daño y la muerte progresivos, especialmente de la oligodendroglia, explicarían que la formación de mielina y de las moléculas que la componen vayan disminuyendo en cantidad a medida que la patología avanza ocasionando la progresión clínica de la enfermedad (6,11,12).
La forma clínica más frecuente de EK es la infantil como es el caso descrito en esta publicación. Se presenta en lactantes de 3 a 6 meses de vida, hasta entonces aparentemente sanos tal como ocurrió en este caso. La edad tiene relación con la importancia de este período en la mielinización del sistema nervioso. La irritabilidad marcada, el llanto persistente, el sobresalto exagerado ante los sonidos y la tendencia al opistótonos son los primeros síntomas que llaman la atención a los padres (6). Pronto se hacen evidentes la falta de adquisición de nuevas conductas, la dificultad para alimentarse y la presencia de vómitos (5) La mayoría de estas manifestaciones se observaron en este caso. La paciente tuvo un período libre de síntomas en el cual cumplió con las pautas del desarrollo de acuerdo a las guías nacionales (13): a nivel motor se sentaba con leve apoyo, en la coordinación tenía prehensión voluntaria, en el área del lenguaje balbuceo imitativo y a nivel del área social: reconocía caras familiares, realizaba juegos con las manos y juegos cara-cara.
Se subraya la de regresión de pautas madurativas (pérdida de hitos de maduración previamente logrados) como hecho fundamental a tener presente por el pediatra para el planteo de una afección neurodegenerativa de base genética. En el caso que se presenta, el pediatra tratante detectó este elemento de alarma así como irritabilidad excesiva y dificultad en la alimentación. Esto motivó derivación a un nivel terciario para completar el diagnóstico y adoptar decisiones terapéuticas. Se destaca la importancia de la vigilancia sistemática del desarrollo del niño y la aplicación periódica de instrumentos de pesquisa que permitan detectar alteraciones en forma oportuna, como en el caso reportado (13).
El bajo incremento ponderal que presentaba es debido en parte al trastorno en la deglución y vómitos que se asocian a la EK, lo que dificulta la alimentación ocasionando el deterioro nutricional progresivo (6).
Dentro de la forma infantil de la EK, se distingue además una variedad de presentación infantil tardía, que se manifiesta en niños entre los 6 meses y los 3 años de vida, también con irritabilidad marcada, ataxia, síndrome piramidal y deterioro psicomotor progresivo (6).
Frente a un cuadro clínico sugestivo de EK la investigación inicial debe incluir el examen del LCR y estudio de neuroimagen. Los estudios iniciales para valoración metabólica básica suelen ser normales (12). A nivel del LCR se puede encontrar en la mayoría de los casos una hiperproteinorraquia, a niveles cercanos a 100 mg/dl como en este caso (6). La resonancia magnética (RM) de cráneo muestra lesiones de atrofia, principalmente donde la mielinización es más temprana. Lesiones simétricas de sustancia blanca con afectación periventricular y atrofia cerebral progresiva también son descritas (7). Las imágenes de atrofia son tardías en la evolución de la enfermedad. Lo más característico en la RM en el primer año de vida es el aumento de la señal en T2 a nivel de la sustancia blanca profunda y periventricular, particularmente en el sector frontal posterior y parietal anterior y de los núcleos profundos y de la sustancia blanca del cerebelo (14).
]]> La RM con espectroscopia permite la detección del compromiso de sustancia blanca antes que ésta sea evidente por la RM convencional. En la EK la espectroscopía presenta en la sustancia blanca elevaciones de los niveles de mioinositol, colina y creatina, junto a la disminución de N-acetilaspartato. En forma inconstante, puede observarse un aumento del pico de lactato (14). En este caso se observó una disminución de la N-acetilaspartato, con aumento de la colina y de los picos de lactato. Estos hallazgos reflejan el proceso de desmielinización, proliferación de las células de la glía y destrucción neuroaxonal (6).Otras alteraciones que se pueden observar es la disminución progresiva de la velocidad de conducción neural periférica y deterioro progresivo de los potenciales visuales auditivos y visuales como se observa en esta publicación.
El diagnóstico de certeza de esta enfermedad se realiza a través de la dosificación de la enzima galactocerebrósido-b-galactosidasa habitualmente en leucocitos, la cual se encuentra disminuida.
El pronóstico de esta patología es muy malo dado que la muerte se produce entre el primer y segundo año de vida. Hasta el momento no se encuentra disponible un tratamiento efectivo que logre evitar el curso progresivo e inexorable de la EK. Se han realizado investigaciones científicas en animales de experimentación por medio de trasplante de células hematopoyéticas troncales, con resultados variables (6). Estos pacientes deben recibir medidas generales tendientes a mejorar la calidad de vida.
Actualmente el objetivo del diagnóstico es para orientación familiar en base a asesoramiento genético, por tratarse de una enfermedad genética con herencia autosómica recesiva, el riesgo de recurrencia es del 25% en cada futura gestación de la misma pareja. Es importante saber que es posible el diagnóstico prenatal mediante el análisis de la actividad de la enzima galactocerebrósido-b-galactosidasa de vellosidades coriales o de cultivos de amniocitos. Para algunos autores, se debería evitar el diagnóstico prenatal basado solamente en el análisis directo de las vellosidades coriales pues la toma de la muestra conlleva el riesgo de contaminación con tejido materno que presenta niveles enzimáticos por sobre los rangos de diagnóstico (6,9). El diagnóstico prenatal también es posible realizando estudio molecular de ADN fetal, pero para ello es indispensable conocer las dos mutaciones en el caso índice o afectado de la familia previo al diagnóstico prenatal (5).
Dado que las enfermedades neurometabólicas son pocos frecuentes, es fundamental su sospecha clínica. Su expresión clínica es muy diversa, pero para el grupo de afecciones lisosomales como es el caso de la EK, el pediatra debe considerar esta posibilidad, ante la presencia de síntomas neurológicos progresivos y persistentes con o sin compromiso de otros sistemas orgánicos. Es relevante la evaluación del desarrollo en el primer nivel de atención dado que algunas de estas enfermedades tienen tratamiento específico como puede ser el transplante de precursores hematopoyéticos o terapia de reemplazo enzimático y mejor pronóstico a largo plazo cuanto más temprano sea el mismo a lo que se suma, en todos los casos, el valor del asesoramiento genético a los padres. Jerarquizamos la importancia de la utilización sistemática de la guía nacional de vigilancia del desarrollo en el niño que sugiere referir al niño cuando se detecten: la falta de uno o más logros en diferentes áreas, presencia de signos de alerta, retroceso o pérdida de uno o más logros adquiridos o si el examen clínico presenta signos de organicidad.
Referencias bibliográficas
1. Krabbe K. A new familial, infantile form of diffuse brain sclerosis. Brain 1916; 39: 74.
]]>2. Austin J, Suzuki K, Armstrong D, Brady R, Bachhawat BK, Schlenker J, et al. Studies in globoid (Krabbe) leukodystrophy (GLD). V. Controlled enzymic studies in ten human cases. Arch Neurol 1970; 23: 502.
3. Suzuki Y, Suzuki K. Krabbe’s globoid cell leukodystrophy Deficiency of galactocerebrosidase in serum, leukocytes, and fibroblasts. Science 1971; 171: 73.
4. Zlotogora J, Regev R, Zeigler M, Iancu TC, Bach G. Krabbe disease Increased incidence in a highlyinbred community. Am J Med Genet 1985; 21: 765.
5. Wenger D, Suzuki K, Suzuki Y, Suzuki K. Galactosylceramide Lipidosis: Globoid Cell Leukodystrophy (Krabbe Disease). In: Scriver CR, Beutler AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York: McGraw-Hill, 2001: 3669-94.
6. Santana M. La enfermedad de Krabbe y la leucodistrofia metacromática. En: Sanjurjo P, Baldellou A. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. 3 ed. Madrid: Ergón; 2009: 639-44.
7. Fejerman N, Fernández Álvarez E. Enfermedad de Krabbe. 3 ed. Buenos Aires: Médica Panamericana, 2007: 364-5.
8. Lyon G. Encefalopatías metabólicas y degenerativas infantiles precoces (1 a 12 meses). En: Neuropediatría. 1 ed. Barcelona: Editorial Masson, 1990: 207-16.
9. Johnston M. Trastornos degenerativos de la infancia. En: Nelson. Tratado de Pediatría. 17 ed. Madrid: Elsevier, 2004: 2029-32.
10. Giachetto G. Mortalidad infantil en Uruguay: una mirada crítica. Arch Pediatr Urug 2010; 81(3): 139-40.
11. Villegas Castrejón H, Hernández-Pérez A, Peralta S, Vázquez Escamilla S, Reyes Marín B. Diagnóstico de leucodistrofia de Krabbe por microscopia electrónica de transmisión. Informe de un paciente. Cir Ciruj 2006; 74: 477-81.
12. Vilanova L, Santos L. Doença de Krabbe (leucodistrofia célula globóide): relato de um caso. J Pediatr (Rio J) 1998; 74 (2): 153-6.
13. Uruguay. Ministerio de Salud Pública. Programa Nacional de Salud de la Niñez. Área Ciclos de Vida. Guía nacional para la vigilancia del desarrollo del niño y de la niña menores de 5 años. Montevideo: MSP, 2010.
14. Barkovich AJ, Patay Z. Metabolic, toxic and inflammatory brain disorders. En: Barkovich AJ, Raybaud C, eds. Pediatric Neuroimaging. 5 ed. Philadelphia: Lippincott Williams & Wilkins, 2012: 81 – 239.
Correspondencia: Dr. Ricardo Tambasco. Correo electrónico: elben82@hotmail.com
]]>{kind=link}