 ]]>
]]>
CASO CLINICO
Arch Pediatr Urug 2009; 80(1)
Síndrome de Steinert neonatal:
distrofia miotónica tipo 1 congénita
Dres. Jorge Arturo Aviña Fierro 1, Daniel Alejandro Hernández Aviña 2
Resumen
El síndrome de Steinert neonatal es una forma severa de distrofia miotónica, caracterizado por hipotonía muscular, dificultades para la alimentación y debilidad de los músculos respiratorios, presentes desde el nacimiento; se hereda en forma autosómica dominante y está causado por la mutación de un gen llamado proteinquinasa de distrofia miotónica (DMPK) localizado a nivel cromosómico 19q locus 13.2, con expansión repetitiva específica de trinucléotidos citosina-timina-guanina (CTG).
Este es un caso de distrofia miotónica congénita en una recién nacida con hipotonía neonatal severa, diplejía facial, problema para la alimentación, dificultad respiratoria y parálisis diafragmática que requirió asistencia ventilatoria con desenlace fatal por insuficiencia respiratoria y muerte al mes de edad. Caso ilustrativo de recién nacido hipotónico donde se enfatizan los criterios clínicos de síndrome Steinert a edad neonatal y su diagnóstico diferencial.
Summary
Neonatal Steinert syndrome is a severe form of myotonic dystrophy, characterized by neonatal muscle hypotonia, feeding difficulties and respiratory muscle weakness. All of them are present since birth; it is inherited as an autosomal dominant trait. It is caused by a mutation in a gene called myotonic dystrophy protein kinase (DMPK) located at chromosome 19q locus 13.2, with a specific repeat expansion of cytosine-thymine-guanine (CTG) trinucleotides. A case of congenital myotonic dystrophy in a female newborn with severe neonatal hypotonia, facial diplegia, respiratory and feeding difficulties and diaphragmatic paralysis is presented. She required respiratory assistance with final death from respiratory failure when she was one month old.
This is an illustrative case of an infant with neonatal hypotonia. The main clinical signs of Steinert syndrome at neonatal age and the differential diagnosis are presented.
Key words: ]]>
MYOTONIC DYSTROPHY-congenital
MUSCLE HYPOTONIA
INFANT, NEWBORN
Introducción
El síndrome de Steinert neonatal, o distrofia miotónica congénita, es un raro padecimiento hereditario autosómico dominante trasmitido por vía materna. El recién nacido afectado manifiesta debilidad, hipotonía, facies miopática, problemas respiratorios y dificultad para la alimentación, además suele tener pie equino (1). La primera descripción clínica detallada la realizó Steinert hace 100 años, en 1909, tres años después Curschmann delineó las manifestaciones neurológicas. Anteriormente se llamaba enfermedad de Steinert, de la cual se conoce la causa, pero se ignora la fisiopatología, siendo así que la denominación correcta es síndrome de Steinert (2). La afección neonatal la reportó Bell en 1972; tiene distribución mundial con incidencia de 1/100.000 recién nacidos vivos, con mortalidad elevada cercana al 50% (3). La causa que determinó Stratton en 1993 es un incremento del número de trinucléotidos tipo CTG (citosina-timina-guanina), por alteración de una región no codificante en el brazo corto del cromosoma 19, locus 13.2 afectando al gen DMPK (myotonic dystrophy protein kinase) con expresión principalmente en músculo cardiaco y sistema músculo-esquelético (4). La secuencia de estos tripletes en el sujeto normal varía de 5 a 30 reiteraciones, los pacientes con este tipo de distrofia tienen repeticiones expandidas por arriba de 2.000.
Caso clínico
Recién nacida producto de segunda gestación a término que cursó sin complicaciones, obtenida por cesárea a causa de polihidramnios; Apgar 7,8; peso al nacimiento 2.700 g, talla 48 cm; padres jóvenes de 20 años de edad ambos, matrimonio no consanguíneo, un hermano de dos años, todos aparentemente sanos. Permaneció hospitalizada en el período neonatal por sufrir severa dificultad para succión y deglución que impedían la alimentación, requiriendo nutrición por sonda nasogástrica, además tenía importantes manifestaciones de dificultad respiratoria y periodos de apnea, necesitando asistencia ventilatoria mecánica con respirador.
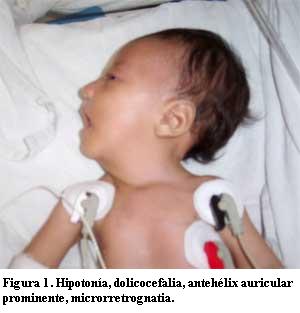
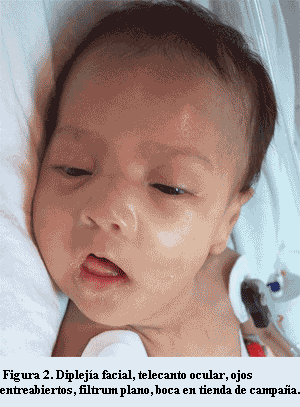
A la exploración física se encontró hipotonía y debilidad generalizada, muy escaso tono y fuerza muscular, cráneo dolicocéfalo, cuello débil sin control de la cabeza por falta de tono flexor, pabellones auriculares grandes con antehélix prominente, mentón caído en retrognatia por hipotonía de maseteros (figura 1). Cara alargada inexpresiva por diplejía facial, telecanto ocular, ojos entreabiertos con incapacidad de oclusión completa; mejillas alargadas, nariz ancha, filtrum plano, boca abierta en forma triangular con labio superior delgado en tienda de campaña por comisuras laterales descendentes; paladar alto ojival, micrognatia (figura 2). Tórax plano longilíneo con pared costal retraída, disminución de ruidos respiratorios en ambos campos pulmonares, área cardíaca con arritmia, taquicardia y soplo sistólico grado II/IV en localización paraesternal izquierda alta. Extremidades con artrogriposis moderada, superiores flexionadas, inferiores hipotónicas con reflejos osteotendinosos disminuidos; contractura de cadera, rodillas y tobillos, artrogriposis moderada, displasia de cadera bilateral, pie equino varo izquierdo.
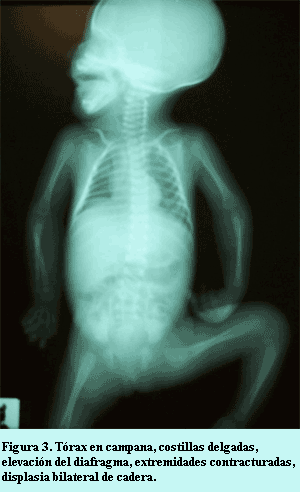
Laboratorio: biometría hemática sin alteraciones, gasometría y electrolitos en niveles normales; química sanguínea: glucosa 70 mg/dL, urea 22 mg/dL, creatinina 0,5 mg/dL, amonio 90 mg/dL, creatinquinasa sérica 350 U/L, tamizaje metabólico negativo. Radiológicamente se encontró tórax en campana, cuerpos costales delgados y finos, silueta cardíaca ovoidea con levantamiento del ápex cardíaco por crecimiento de ventrículo derecho, situación elevada del diafragma y sombra hepática incrementada; extremidades superiores mostrando flexión moderada de mano sobre antebrazo y brazo en ambos lados, y las inferiores con flexión de pierna sobre muslo, displasia de cadera bilateral (figura 3). El electrocardiograma mostró ritmo sinusal, frecuencia de 140 por minuto, eje 120º, crecimiento de ventrículo derecho, y el ecocardiograma con hipertrofia leve del tabique ventricular sin obstrucción del tracto de salida del ventrículo izquierdo, comunicación interauricular pequeña tipo foramen oval. La paciente tuvo evolución desfavorable complicándose con convulsiones tónicoclónicas generalizada causadas por encefalopatía hipóxico-isquémica, la tomografía de encéfalo mostró edema cerebral difuso e hipodensidades en corteza cerebral; finalmente presentó arritmia y falla cardiaca congestiva venosa; murió por falla respiratoria severa al mes de edad.
Discusión
El enfoque diagnóstico de la hipotonía se realizó como una miopatía congénita, por ser un problema rápidamente progresivo presente desde el nacimiento, afectando principalmente el sistema muscular y con evolución prolongada, se descartó miopatía metabólica (glucogenosis) o citopatía mitocondrial (encefalomiopática) por no existir acidosis metabólica; tampoco correspondía a miopatía estructural (miotubular o nemalínica) pues existían niveles altos de creatinquinasa (5); de las distrofias musculares, se descartaron la forma congénita clásica por deficiencia de merosina, pues no creatinquinasa); el tipo Walker-Warburg (faltaba microftalmía y lisencefalia), el tipo japonés de Fukuyama (carecía de hipoplasia cerebral) y el tipo finlandés de Santavuori (no existía microgiria) (6).
La posibilidad diagnóstica se redujo a distrofia miotónica que es la forma más frecuente de las distrofias musculares, con una prevalencia de 1/20.000 individuos adultos; se caracteriza por debilidad progresiva muscular (distrofia) y espasmos musculares o rigidez con dificultad para el relajamiento muscular después de contracciones repetidas (miotonía). La afección congénita es una enfermedad sistémica grave que puede afectar ambos sexos, y se presenta en hijos de madres con distrofia manifiesta o sutil (7); resulta difícil de diagnosticar durante la etapa neonatal, debiendo constatarse los criterios clínicos de Wesstrom: hipotonía, facies miopática, pobre control de la cabeza, debilidad y atrofia muscular, problemas para la alimentación y dificultad respiratoria (8); existen, además, otros hallazgos complementarios como son: parálisis diafragmática, costillas delgadas y deformación esquelética como pie equino (9). El caso tenía clínicamente todos los datos (10) y además la madre, aunque no mostraba sintomatología de la enfermedad, cuando se le ordenaba abrir y cerrar el puño en forma repetida o sacudir las manos presentaba respuesta fatigada, orientadora de distrofia miotónica subclínica. Los padres no permitieron biopsia muscular de la paciente que hubiera identificado fibras en anillo con miofilamentos periféricos, núcleos en disposición central y masas sarcoplásmicas. Se dio consejo genético, pues es un problema hereditario que muestra anticipación, que expresará mayor severidad fenotípica en hijos subsecuentes, siendo la posibilidad de recurrencia mayor del 50% en cada nuevo embarazo (11).
]]>Referencias bibliográficas
1. Roig M, Balliu PR, Navarro C, Brugera R, Losada M. Presentation, clinical course, and outcome of the congenital form of myotonic dystrophy. Pediat Neurol 1994; 11: 208-13.
2. Steinberg H, Wagner A. Hans Steinert: 100 years of myotonic dystrophy. Nervenarzt 2008; 79: 961-70.
3. Bell DB, Smith DW. Myotonic dystrophy in the neonate. J Pediatr 1972; 81: 83-6.
4. Stratton RF, Patterson RM. DNA confirmation of congenital myotonic dystrophy in non-immune hydrops fetalis. Prenat Diagn 1993; 13: 1027-30.
]]>5. Dalphin ML, Noir A, Monnier G, Menget A. Congenital myotonic dystrophy. Diagnostic difficulties. Pediatrie 1992; 47: 677-80.
6. Klein A, Clement E, Mercuri E, Muntoni F. Differential diagnosis of congenital muscular dystrophies. Eur J Paediatr Neurol 2008; 12: 371-7.
7. Schara U, Schoser BG. Myotonic dystrophies type 1 and 2: a summary on current aspects. Semin Pediatr Neurol 2006; 13: 71-9.
8. Wesstrom G, Bensch J, Schollin J. Congenital myotonic dystrophy. Incidence, clinical aspects and early prognosis. Acta Paediatr Scand 1986; 75: 849-54.
9. Yong SC, Boo NY, Ong LC. A case of congenital myotonic dystrophy presented with diaphragmatic paresis during the neonatal period. J Paediatr Child Health 2003; 39: 567-8.
]]>10. De León MB, Cisneros B. Myotonic dystrophy 1 in the nervous system: from the clinic to molecular mechanisms. J Neurosci Res 2008; 86: 18-26.
11. Lesca G, Hays S, Bourgeois J, Bost M, Ollagnon-Roman E, Putet G. Diagnosis of congenital myotonic dystrophy in a neonate: its familial consequences. Arch Pediatr 2003; 10: 466-7.
Correspondencia: Dr. Jorge Arturo Aviña Fierro.
Alberto Cossío 1432, Huentitán El Alto. Guadalajara, México
Correo electrónico: avinafie@megared.net.mx
]]>