Neoplasia endocrina múltiple tipo 1. Presentación de una familia afectada con diagnóstico molecular
Dres. Mariela Larrandaburu*, Alicia Vaglio†,
Andrea Quadrelli‡, Roberto Quadrelli§
Instituto de Genética Médica, Hospital Italiano. Montevideo, Uruguay
La neoplasia endocrina múltiple tipo 1 (MEN1) incluye una combinación variable de más de 20 tumores endocrinos y no endocrinos, con herencia mendeliana autosómica dominante. El paciente, con antecedentes familiares de neoplasias, consultó a los 39 años con historia de tumor mediastinal y neoplasma de cabeza de páncreas. Se realizó diagnóstico clínico de MEN1 y aceptó ser incluido en un estudio cooperativo conducido por el grupo francés para el estudio de MEN. Se realizó la secuenciación del gene MEN1 detectándose una mutación puntual en el exon 10: 1596delA. Se le informó de la relevancia de realizar el mismo estudio en otros integrantes de la familia en riesgo de presentar igual patología. Dos años después, consultó la hermana de 39 años con historia de hiperparatiroidismo, calciuria, hormona paratiroidea (PTH) elevada, litiasis renal y operada de adenoma paratiroideo. Refiere que el propósito falleció. Dado el antecedente familiar, se estudió específicamente la mutación previamente encontrada en el hermano, confirmando el diagnóstico de MEN1. La consultante decidió conocer el estatus molecular de sus dos hijas asintomáticas, no detectándose la mutación en ninguna de ellas. Este trabajo ilustra el beneficio de un diagnóstico que requiere tecnología de alto costo (secuenciación) a través de un estudio colaborativo y que permite que otros integrantes de la familia puedan conocer su definición molecular. Destacamos la importancia del asesoramiento genético en la toma de decisión de realización del diagnóstico molecular en individuos asintomáticos y la jerarquía del diagnóstico precoz para definir el protocolo de seguimiento en este grupo de pacientes.
Palabras clave: NEOPLASIA ENDOCRINA MÚLTIPLE TIPO 1.
CROMOSOMAS HUMANOS PAR 11.
Key words: MULTIPLE ENDOCRINE NEOPLASIA TYPE 1.
CHROMOSOMES, HUMAN, PAIR 11.
* Genetista. Magíster en Genética y Biología Molecular. Universidad Federal de Río Grande del Sur (UFRGS), Brasil.
]]> † Genetista. Subdirectora del Instituto de Genética Médica, Hospital Italiano. Uruguay.‡ Licenciada en Biología. Magíster en Antropología Social. Doctora en Antropología Social (UFRGS), Brasil.
§ Director del Instituto de Genética Médica. Miembro titular de la Academia Nacional de Medicina. Uruguay.
Correspondencia: Dra. Mariela Larrandaburu
Instituto de Genética Médica, Hospital Italiano.
Bulevar Artigas 1632, CP 11600. Montevideo, Uruguay.
www.geneticamedica.com.uy
Correo electrónico: institutodegenetica.mariela@gmail.com ; rquadr@dedicado.net.uy
Recibido: 12/5/08.
Aceptado: 21/7/08.
]]>Introducción
La neoplasia endocrina múltiple tipo 1 (OMIM 131100), conocida por su sigla en inglés MEN1 (Multiple Endocrine Neoplasia, type 1), también denominada adenomatosis endocrina múltiple tipo I o síndrome de Wermer, es una enfermedad caracterizada por la aparición de tumores glandulares, generalmente benignos, en glándulas endocrinas como la paratiroides, hipófisis y páncreas(1). Con menor frecuencia se describe la presencia de lipomas viscerales y cutáneos, carcinoides bronquiales e intestinales, adenomas tiroideos, bocios difusos y neoplasias de la corteza adrenal(1-3). El hiperparatiroidismo es la alteración más frecuente y aparece en más de 90% de los pacientes; entre 30% y 75% de los pacientes padecen tumores pancreáticos que producen insulina u otras hormonas pancreáticas como gastrina (una de las más frecuentes) provocando la aparición de múltiples úlceras de estómago(4,5). La prevalencia de la enfermedad se estima en aproximadamente uno cada 30.000 individuos y los síntomas de la enfermedad generalmente comienzan en la cuarta o quinta década de vida(3,6,7).
La enfermedad se hereda como un rasgo genético autosómico dominante y es causada por mutaciones en el gene MEN1 localizado en el brazo largo (q13) del cromosoma 11(8,9). El gene MEN1 es un gene supresor tumoral con 10 exones y una región codificante de 1.830 pares de bases que codifica una proteína de 610 aminoácidos denominada menin(10). Se han identificado alrededor de 300 mutaciones germinales en pacientes con MEN1 y aproximadamente 25% de éstas son mutaciones sin sentido, 45% deleciones, 15% inserciones, menos del 5% son mutaciones que producen defectos en el splicing y 10% mutaciones que producen una pérdida de sentido en la lectura del código genético(1,11). La mayoría de estas mutaciones (>80%) producen inactivación del gene, lo cual es consistente con lo esperado en un gene supresor tumoral(1). El diagnóstico de MEN1 se inicia con la sospecha clínica en pacientes que presentan enfermedad de úlcera péptica, síntomas relacionados con hipoglicemia, hipercalcemia, hipercalciuria o litiasis renal, o ambas, así como síntomas relacionados con problemas pituitarios. El diagnóstico clínico de MEN1 esporádico se realiza por la presencia de dos de tres tumores endocrinos, a saber: tumor de paratiroides, tumor pituitario y tumor diferenciado del tracto gastro-entero-pancreático. El diagnóstico clínico de MEN1 familiar se realiza: a) en aquellos individuos con antecedentes de un pariente en primer grado con uno de los tres tumores endocrinos requeridos para el diagnóstico; b) en individuos con un órgano involucrado y la detección de la mutación germinal MEN 1(12,13).
El MEN1 puede desarrollarse muy lentamente, por tal motivo la descendencia no debe ser excluida como posibles portadores hasta la edad de 35 años, a pesar de determinaciones bioquímicas normales(3). Además, al no ser lo más apropiado la cirugía preventiva en muchos de los órganos con mayor riesgo de desarrollo de tumores (como duodeno, páncreas y pulmones), se recomienda que las personas con riesgo de MEN1 sean monitoreadas de rutina con análisis bioquímicos e imagenológicos. Dichos análisis pueden detectar la aparición de la enfermedad cerca de diez años antes del desarrollo de los síntomas permitiendo, por tanto, un tratamiento preventivo(3).
Los pacientes con MEN1 tienen un mayor riesgo de muerte prematura. En los primeros estudios el promedio de vida de los pacientes era de alrededor 50 años, constituyendo la principal causa de muerte por úlcera péptica y complicaciones subsecuentes causadas por gastrinoma e hiperparatiroidismo(12). El tratamiento médico de las úlceras pépticas ha mejorado mucho y ha disminuido el riesgo de muerte prematura; hoy día, el determinante más significativo en relación con el pronóstico de pacientes con MEN1 es la transformación maligna de tumores gastroenteropáticos y tumores carcinoides tímicos/bronquiales(12).
El síndrome MEN1 es monogénico y, de acuerdo con su patrón de herencia dominante; cada paciente afectado tiene una probabilidad de 50% de transmitir el gene mutado a su progenie, independientemente del sexo. El asesoramiento genético es muy importante en las familias afectadas. En la actualidad es posible identificar prematuramente a las personas afectadas o con riesgo de ser portadoras por estudios genéticos(13). Los estudios moleculares de secuenciación directa de regiones codificantes permiten detectar mutaciones en, al menos, 85% de las familias afectadas. La identificación de la mutación en una familia afectada permite detectar su presencia en personas sin síntomas clínicos y con riesgo de ser portadoras; en este último caso, se indica la realización de estudios bioquímicos o radiológicos, o ambos, en forma periódica. Por otra parte, se observa una enorme variabilidad en relación con la localización y manifestación de los tumores asociados al MEN1, y también en relación con el comportamiento clínico aun en pacientes que comparten la misma mutación MEN1(3). Por esta razón, es muy difícil prever un determinado fenotipo clínico en individuos portadores asintomáticos. Así como las manifestaciones clínicas del MEN1 son muy variables, inclusive dentro de una misma familia, también lo son la edad de aparición y su evolución. La falta de correlación genotipo-fenotipo sugiere el posible rol de otros genes o factores ambientales, o ambos(3).
En este trabajo presentamos una familia uruguaya con confirmación bioquímica, imagenológica y molecular de MEN1. Se estudiaron cuatro integrantes de la familia (dos hermanos y dos hijas de uno de éstos). Destacamos la importancia del diagnóstico precoz de la enfermedad para instrumentar un tratamiento preventivo antes de la aparición de los primeros síntomas, y la definición molecular que permite el diagnóstico y el asesoramiento genético a todos los integrantes de la familia, incluyendo también el diagnóstico prenatal.
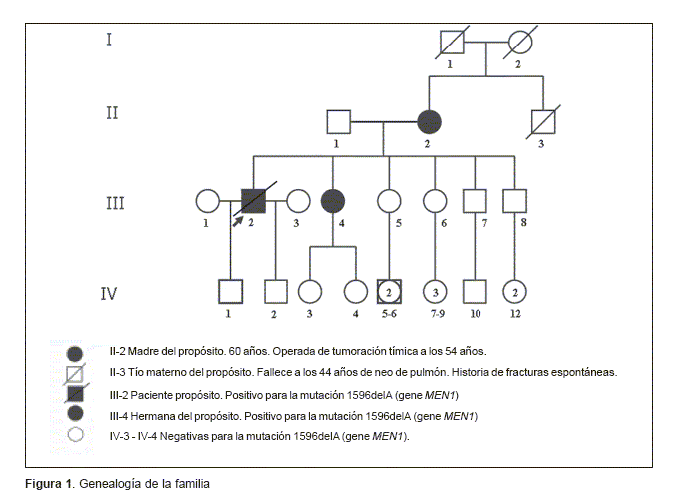
]]> Material y métodoCaso clínico 1 (individuo III-2, figura 1)
Caso propósito: paciente de sexo masculino de 39 años al momento de la consulta en el Instituto de Genética Médica del Hospital Italiano. Con historia de múltiples cirugías de tejido glandular, a los 34 años se interviene de una tumoración torácica (masa de mediastino superior), con diagnóstico anatomopatológico de timoma fusocelular. Posteriormente, es reintervenido de un tumor carcinoide de timo atípico. Cuatro años más tarde presenta: ictericia obstructiva y tumoración pancreática, con diagnóstico operatorio de neoplasia de cabeza de páncreas con múltiples adenomegalias. La anatomía patológica indicó metástasis ganglionar de neoplasia epitelial de histogénesis neuroendocrina.
Con relación a los antecedentes familiares, se describieron antecedentes de tumoración tímica en la madre (II-2), y un tío materno (II-3), quien falleció a los 44 años de probable neoplasma de pulmón, con historia de fracturas espontáneas (figura 1).
Con estos antecedentes, la familia realizó interconsulta con la Dra. Elisabeth Brambilla, del Laboratorio de Patología Celular de Grenoble (Francia), quien sugirió el diagnóstico de MEN1 y la realización del estudio genético molecular. Con este diagnóstico el paciente consultó en nuestro instituto, donde se implementó el estudio de genética molecular que incluyó el análisis del gene para MEN1 en colaboración con el Dr. Alain Calender, del Hospital Edouard Herriot (Lyon Cedes, Francia). Brevemente, se extrajo ADN de la sangre y se realizó secuenciación directa de los exones 2-10 del gene MEN1 para identificar posibles mutaciones según técnicas estándar descriptas previamente(14,15).
Se confirmó el diagnóstico propuesto, al identificarse una mutación germinal en el exón 10: 1596delA del gene MEN1. Se realizó asesoramiento genético individual que, conceptualmente, se define como un proceso de comunicación en términos inteligibles para el interlocutor respecto de la patología, su pronóstico y manejo disponible, respondiendo a las inquietudes del paciente(16). Considerando el mecanismo de herencia de la enfermedad, el asesoramiento implica la sugerencia de búsqueda de la mutación en los integrantes de la familia que, genealógicamente, presentan riesgo de ser portadores, lo que permitiría la realización de asesoramiento genético familiar. Teniendo en cuenta aspectos bioéticos, se informa al paciente que la decisión del estudio molecular para conocer si es portador de la mutación, con probable expresión clínica y potencial transmisor de la misma a su descendencia, es privativa de cada uno de los familiares.
Los integrantes del Instituto de Genética Médica mantuvieron contacto telefónico con el paciente, cuyo seguimiento clínico fue realizado por médico endocrinólogo. La severidad de su condición clínica determinó el fallecimiento un año más tarde, a los 40 años de edad.
]]> Caso clínico 2 (individuo III-4, figura 1)Hermana del propósito. Se trata de una mujer de 39 años al momento de la consulta en el Instituto de Genética Médica del Hospital Italiano, producto de segundo embarazo y parto de pareja no consanguínea de 21 años la madre y 31 el padre al momento del nacimiento (figura 1). Concurrió por primera vez al Instituto de Genética solicitando asesoramiento genético. Con historia de bocio difuso, eu-funcionante, tratado con hormonas tiroideas (T4) y betabloqueantes, picos hipertensivos y taquicardias en algunas oportunidades. Fractura de húmero izquierdo con evolución tórpida del callo óseo. Cuadro depresivo, que se manifiesta luego del fallecimiento del hermano con diagnóstico genético de MEN1. Cólicos nefríticos a repetición, litiasis renal bilateral con colocación de catéter doble J, por estenosis ureteral derecha, secundario a litiasis. Hiperparatiroidismo.
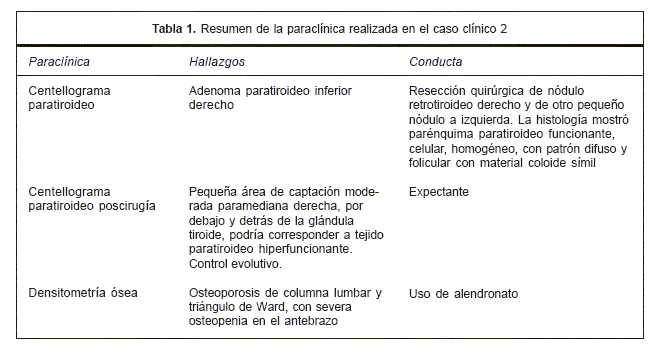
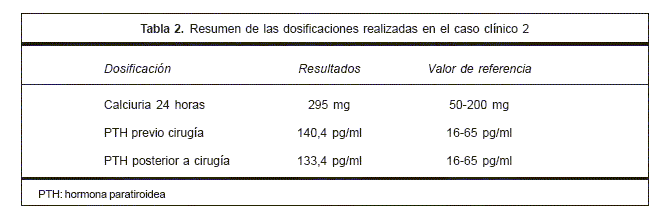
En las tablas 1 y 2 se resume la paraclínica realizada y el tratamiento instrumentado.
Una vez informada de la probabilidad de ser portadora, la paciente decidió realizar el estudio genético molecular para MEN1, manifestando especialmente la preocupación de transmitir la enfermedad a sus hijas. Al conocerse la mutación presente en la familia, se realizó secuenciación directa solamente del exón relevante, es decir, el exón 10 del gene MEN1. El estudio molecular confirmó el diagnóstico de neoplasia endocrina múltiple tipo I. Con este resultado se realizó asesoramiento genético a la paciente y se le sugirió la realización de estudios para determinar la extensión lesional de la patología. Una vez asesorada sobre la implicancia de realizar el estudio genético para el gene MEN1 en personas asintomáticas adolescentes, la paciente resuelve efectuar dicho estudio en sus dos únicas hijas, determinándose el estatus de no portadoras de la mutación encontrada en su familia. Estos resultados, que descartan la herencia de la mutación en sus hijas, fueron recibidos con gran alegría por la paciente. Actualmente es evaluada periódicamente para despitar precozmente manifestaciones clínicas de la enfermedad y aplicar el tratamiento correspondiente.
Resultados
En el caso clínico 1 se identificó una mutación en el exon 10 del gene MEN1 referida como 1596delA que confirmó el diagnóstico de MEN1. Se realizó la búsqueda de la mutación puntual en la hermana del propósito (caso clínico 2) donde se identificó igual mutación. Posteriormente, dicha paciente decidió realizar el estudio molecular en sus dos hijas, que resultó negativo, es decir, no portadoras de la mutación 1596delA detectada en su madre y en uno de sus tíos.
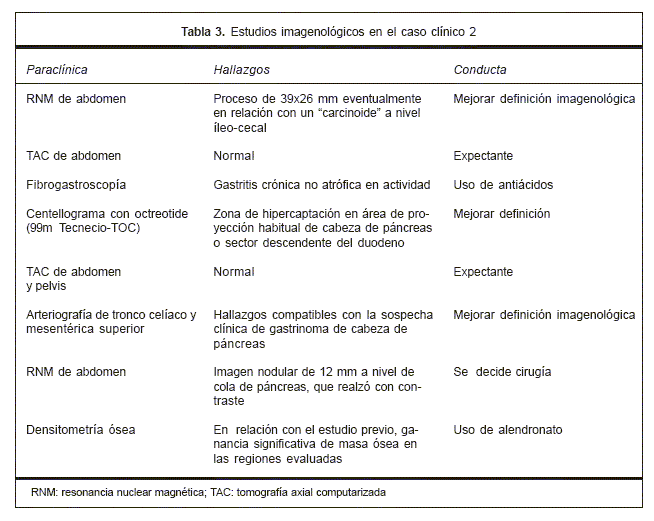
Se realizó seguimiento de la paciente (caso clínico 2) hasta la actualidad. El enfoque de la patología incluye diferentes especialistas (endocrinólogos, gastroenterólogos, cirujanos, entre otros). Los estudios imagenológicos, anatomopatológicos y bioquímicos más relevantes están sintetizados en las tablas 3 y 4, respectivamente. La paciente fue intervenida quirúrgicamente con diagnóstico preoperatorio de gastrinoma múltiple pancreático, realizándose una duodenopancreatectomía cefálica (DPC) con resección de bazo accesorio, vesícula biliar y adenopatía gastroepiploica. El informe anatomopatológico refiere: presencia de 25 tumores endócrinos pancreáticos histológicamente benignos, un microcarcinoide intramucoso gástrico y un bazo accesorio.
]]>Discusión
El síndrome MEN1 se caracteriza por la aparición combinada de tumores endocrinos de paratiroides, células de los islotes del páncreas y pituitaria anterior. Los tumores paratiroideos se observan en 95% de los pacientes con MEN1 con la manifestación de hipercalcemia causada por hiperparatiroidismo primario(1). Los tumores paratiroideos detectados por hipercalcemia constituyen la primera manifestación de MEN1 en cerca de 90% de los pacientes. Así lo fueron en los casos clínicos presentados en este trabajo. Los gastrinomas, que provocan la aparición del síndrome de Zollinger-Ellison, son la causa más importante de morbimortalidad en los pacientes con MEN1. Este hallazgo fue confirmado en el caso clínico 2, del punto de vista bioquímico y anatomopatológico. Por otra parte, la historia clínica del propósito fue señalada por el Dr. Calender como "llamativa" en el sentido de una presentación atípica (con formas atípicas de timoma) de la enfermedad de MEN1 (comunicación personal).
Una vez realizado el diagnóstico de MEN1, el protocolo de seguimiento de los pacientes afectados incluye: 1) exploraciones de las glándulas endocrinas clásicamente afectadas en el síndrome: paratiroides, páncreas, pituitaria, glándulas adrenales, además de las lesiones tímicas características; 2) detección y el tratamiento de gastrinoma y de otros tumores entero-hepáticos neuroendocrinos, feocromocitoma, prolactinoma, uso de agentes antirreabsortivos de hueso (para reducción de hipercalcemia), eventual paratiroidectomía, detección y tratamiento de tumores hipofisarios secretantes y no secretantes; 3) diagnóstico y tratamiento de las eventuales complicaciones posquirúrgicas como el desarrollo de diabetes secundaria a la resección pancreática(12).
Los síndromes MEN son poco frecuentes, pero al ser heredados como rasgos autosómicos dominantes, su diagnóstico tiene gran importancia para otros miembros de la familia, pues los parientes en primer grado tienen 50% de riesgo de desarrollar la enfermedad. En los integrantes de la familia con 50% de riesgo de tener el síndrome MEN1, y cuya definición genética es desconocida, está indicado realizar determinaciones bioquímicas específicas que incluyen dosificación de prolactina, calcio sérico, PTH y gastrina. La oportunidad de la realización de estudios moleculares en los integrantes de la familia en situación de riesgo deberá ser siempre precedida del asesoramiento genético respectivo por las implicancias psicológicas potenciales al realizarse estudios diagnósticos en individuos presintomáticos. A los 20 años, aproximadamente, la mitad de los pacientes ya presentan algún síntoma y a los 40, 98% de los mismos son sintomáticos(17).
En estos últimos años se han realizado importantes avances en los síndromes MEN, desde su reconocimiento clínico, en una primera etapa, hasta la identificación de los genes involucrados y sus defectos moleculares, que incluyen la definición de algunas de las funciones de las proteínas codificadas(1). Sin embargo, se observa una gran diversidad en el tipo de mutaciones observadas y en su distribución en la región codificante del gene MEN1. Algunas mutaciones (que representan alrededor de 25% de todas las mutaciones observadas) son más frecuentes que otras y están presentes en familias no relacionadas(1). También se observa una falta de correlación entre las mutaciones del gene MEN1 y las manifestaciones clínicas de la enfermedad, a diferencia de lo observado en los síndromes MEN tipo 2(1). Hasta donde sabemos, la mutación identificada en la familia aquí presentada no ha sido reportada previamente en la literatura.
Por otra parte, según se mencionó antes, el gene MEN1 codifica una proteína supresora de tumor denominada menin cuya función bioquímica se desconoce; si bien, recientemente, se ha identificado a esta proteína como un regulador de la transcripción génica, de la proliferación celular, apoptosis y estabilidad genómica(11). Estos descubrimientos sugieren que las diversas funciones de la proteína dependen de su asociación con la cromatina y su control sobre la transcripción génica(11). Sin embargo, su rol preciso en el núcleo y en la regulación del control del crecimiento celular endocrino aún se desconoce(1). En los pacientes que aquí se describen, el efecto funcional de la mutación encontrada, referida como 159delA y localizada en el exón 10 del locus MEN1, produce una pérdida en el sentido de lectura del código genético induciendo un codón STOP prematuro que origina una proteína menin truncada, probablemente inestable en la célula.
A modo de conclusión, como indicamos antes, el enfoque de esta patología exige la colaboración conjunta de diferentes especialistas para un mejor manejo de los pacientes afectados y sus familias; como plantea Marini y colaboradores, el objetivo principal para los pacientes con MEN1 es ofrecerles un óptimo programa de tratamiento y de prevención de cáncer(3). Destacamos la importancia del diagnóstico precoz de la enfermedad para instrumentar un tratamiento preventivo antes de la aparición de los primeros síntomas. La definición molecular permite el diagnóstico y asesoramiento genético a todos los integrantes de la familia en cuanto a la posibilidad de presentar la mutación y desarrollar la enfermedad, así como de transmitirla a su descendencia, pudiendo realizarse diagnóstico prenatal.
]]> AgradecimientosA los diferentes especialistas que han realizado estudios paraclínicos y tratamiento específico tanto en Paysandú como en Montevideo: Prof. Dra. Cristina Belzarena, Dra. Alba Echeverrigaray, Dr. Parodi, Dr. Pignata, Dra. Silvia Episcopo, Dr. Gonzalo Ardao, Dr. Raúl Lozán, Dr. Jorge Crosa, Dr. Antonio Páez, Dr. Javier Vilar, Dr. Rodolfo Ferrando. Así como a la Dra. Elisabeth Brambilla, del Laboratorio de Patología Celular de Grenoble (Francia), y al Dr. Alain Calender del Hospital Edourd Herriot (Lyon Cedes, Francia) por su colaboración en el presente estudio. Agradecemos especialmente a los pacientes y a sus familiares por su amable participación y contribución a este trabajo.
Summary
Multiple endocrine neoplasia type 1(MEN1) comprises a variable combination of over 20 endocrine and non-endocrine tumors, with Mendelian autosomal dominant inheritance. The patient, who had a family history of tumors, was seen, at age 39, due to a mediastinal tumor and neoplasm at the head of the pancreas. We performed clinical diagnosis of MEN1, and we had the collaboration of a French team involved in MEN1 research. We conducted sequenciation of the MEN 1 gene and identified a single mutation in exon 10: 1596delA. The patient was informed about the relevance of performing the same study in other members of the family who were likely to carry the same pathology. Two years later, his 39-year-old sister visited the clinic, with a history of hyperparathyroidism, calciuria, high parathyroid hormone (PTH), renal lithiasis, and who had undergone parathyroid adenoma surgery. She informed her brother had died. Given the family history, we especially studied the mutation previously found in her brother, and confirmed the MEN1 diagnosis. Thus, the patient decided to learn about the molecular status in her two asymptomatic daughters, whereby no mutations were identified. The present study illustrates the benefit of diagnosis, requiring high cost technology (sequenciation) through a collaborative study, which enables other members of a family to learn about their molecular definition. We stress the importance of genetic counseling to make a decision regarding the conduction of molecular diagnosis in asymptomatic individuals and the relevance of early diagnosis to define the follow-up protocol for these patients.
Résumé
La néoplasie endocrine multiple type 1(NEM1) comprend un éventail de plus de 20 tumeurs endocrines et pas endocrines, à héritage mendélien autosomique dominant. Le patient de 39 ans, dont la famille avait des antécédents de néoplasies, portait une histoire clinique de tumeur médiastin et néoplasme de la tête du pancréas. On a abouti au diagnostic clinique de NEM1 et il a accepté d’être inclus dans un travail coopératif mené à bout par une équipe française qui étudie les NEM. On fit la séquence du gène NEM1, une mutation ponctuelle à l’exon 10 fut repérée: 1596delA. On lui a expliqué l’importance de réaliser la même étude à d’autres intégrants de la famille risquant de subir la même pathologie. Deux ans plus tard, la soeur de 39 ans a consulté, avec une histoire d’ hyperparathyroïdie, calciurie, hormone parathyroïde (PTH) élevée, lithiase rénale et une opération d’adénome parathyroïde. Elle confirme que le propos avait décédé. Vu l’antécédent familial, on a étudié spécifiquement la mutation trouvée au préalable chez le frère, confirmant ainsi le diagnostic de NEM1.La patiente décide alors de connaître le statut moléculaire de ses deux filles asymptomatiques, la mutation n’étant repérée chez aucune d’elles. Ce travail illustre le bénéfice d’un diagnostic qui nécessite d’une technologie chère (séquenciation), d’une étude collaborative qui permet à d’autres membres de la famille de connaître leur définition moléculaire. On signale l’importance des données génétiques à la prise de décision de réalisation du diagnostic moléculaire chez des individus asymptomatiques et la hiérarchie du diagnostic précoce afin de définir le protocole de suivi de ce groupe de patients.
]]>Resumo
A neoplasia endócrina múltipla tipo 1 (MEN1) inclui uma combinação variável de mais de 20 tumores endócrinos e não endócrinos com herança mendeliana autossômica dominante. O paciente, com antecedentes familiares de neoplasmas, consultou aos 39 anos com história de tumor mediastinal e neoplasma de cabeça de pâncreas. Foi realizado o diagnóstico clínico de MEN1 e o paciente concordou em ser incluído em um estudo cooperativo conduzido por um grupo francês de estudo das MEN. Na secuenciaçao do gen MEN1 detectou-se uma mutação pontual no éxon 10: 1596delA. O paciente foi informado da importância de realizar o mesmo estudo em outros integrantes da família com risco de apresentar uma patologia similar. Dois anos depois, sua irmã com 39 anos, consultou apresentando hiperparatiroidismo, calciúria, hormônio paratireóideo (PTH) elevado, litiase renal e operada de um adenoma paratiroideo. A paciente relata o falecimento de seu irmão. Considerando o antecedente familiar, a mutação apresentada pelo irmão foi estudada, o que confirmou o diagnóstico de MEN1. A paciente decidiu estudar o estado molecular de suas duas filhas assintomáticas, nas quais não se detectou mutação. Este trabalho mostra o beneficio de um diagnóstico que requer tecnologia de alto custo (sequenciação) através de um estudo colaborativo que permite a outros integrantes da família conhecer sua definição molecular. Destacamos a importância do assessoramento genético para a tomada de decisão na realização de diagnóstico molecular em indivíduos assintomáticos e a importância do diagnóstico precoce para definir o protocolo de seguimento neste grupo de pacientes.
Bibliografía
1. Thakker RV. Multiple endocrine neoplasia–syndromes of the twentieth century. J Clin Endocrinol Metab 1998; 83: 2617-20.
2. Lips CJ, Vasen HF, Lamers CB. Multiple endocrine neoplasia syndromes. Crit Rev Oncol Hematol 1984; 2(2): 117-84.
3. Marini F, Falchetti A, Del Monte F, Carbonell Sala S, Gozzini A, Luzi E, et al. Multiple endocrine neoplasia type 1. Orphanet J Rare Dis 2006; (2):1-38.
4. Lamers CB, Froeling PG. Clinical significance of hyperparathyroidism in familial multiple endocrine adenomatosis type I (MEA I). Am J Med 1979; 66(3): 422-4.
5. Oberg K, Wälinder O, Boström H, Lundqvist G, Wide L. Peptide hormone markers in screening for endocrine tumors in multiple endocrine adenomatosis type I. Am J Med 1982; 73(5): 619-30.
6. Guadagna M, Migliano M, Herrera J, Rodríguez P, Sciorra J, Alfieri A, et al. Diagnóstico molecular de neoplasia endocrina múltiple tipo 1 (MEN-1) en una familia argentina afectada. Medicina (Buenos Aires) 1998; 58: 441-5.
7. Shepherd JJ. The natural history of multiple endocrine neoplasia type 1. Highly uncommon or highly unrecognized? Arch Surg 1991; 126(8): 935-52.
8. Wermer P. Endocrine adenomatosis and peptic ulcer in a large kindred. Inherited multiple tumors and mosaic pleiotropism in man. Am J Med 1963; 35: 205-12.
9. Calender A, Giraud S, Cougard P, Chanson P, Lenoir G, Murat A, et al. Multiple endocrine neoplasia type 1 in France: clinical and genetic studies. J Intern Med 1995; 238(3): 263-8.
10. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997; 276: 404-7.
11. Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol 2007; 265-266: 34-41.
12. Sakurai A, Katai M, Yamashita K, Mori J, Fukushima Y, Hashizume K. Long-term follow-up of patients with multiple endocrine neoplasia type 1. Endocrine J 2007; 54(2): 295-302.
13. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86: 5658-71.
14. Klein RD, Salih S, Bessoni J, Bale AE. Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 2005; 7: 131-8.
15. Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjöld M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab 2007; 92: 3389-95.
16. Rantanen E, Hietala M, Kristoffersson U, Nippert I, Schmidtke J, Sequeiros J, et al. What is ideal genetic counselling? A survey of current international guidelines. Eur J Hum Genet 2008; 16: 445-52.
17. Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 1998; 62: 232- 44.
]]>
{kind=link}
{kind=link}
{kind=link}
{kind=link}