
Aciduria glutárica tipo I
Descripción del primer caso clínico nacional
Dres. Ana Barreiro*, Andrea Rey†, Gabriel González‡,
Aída Lemes§, Álvaro Galiana¶, Leopoldo Peluffo††
Clínica Pediátrica C, Cátedra de Neuropediatría.
Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Resumen
]]> Presentamos el primer caso nacional de un niño portador de una aciduria glutárica tipo I.Esta afección es un error congénito del metabolismo de los aminoácidos esenciales lisina y triptofano determinado por la deficiencia de la enzima mitocondrial glutaril-coenzima A deshidrogenasa, y en nuestro paciente se presenta como la forma clásica de la enfermedad con síntomas neurológicos de instalación aguda que involucran principalmente el sistema extrapiramidal. El diagnóstico se realiza al evidenciar la presencia del ácido 3-hidroxiglutárico, específico de esta afección. El tratamiento está dirigido a evitar el compromiso neurológico en los casos asintomáticos ya que una vez instalado el daño neurológico es irreversible.
Palabras clave: ERRORES INNATOS DEL METABOLISMO DE LOS AMINOÁCIDOS.
* Residente de Pediatría. Clínica Pediátrica C.
† Asistente de Neuropediatría.
‡ Prof. Adjunto de Neuropediatría.
§ Médica. Instituto de Genética Médica. Hospital Italiano.
¶ Ex Prof. Adjunto Clínica Pediátrica C, Servicio Infecto Contagioso.
†† Ex Profesor Clínica Pediátrica C.
]]> Correspondencia: Dra. Andrea ReyConcepción del Uruguay 1870, CP 11400. Montevideo, Uruguay.
E-mail: andrearey@adinet.com.uy
Recibido: 13/11/03.
Aceptado: 17/9/04.
AGI: aciduria glutárica tipo I
GD: enzima mitocondrial glutaril-coenzima A deshidrogenasa
TGO: transaminasas glutámico oxalacéticas
TGP: transaminasas glutámico pirúvica
]]> LDH: lactato deshidrogenasaLCR: líquido cefalorraquídeo
EEG: electroencefalograma
TAC: tomografía axial computarizada
RNM: resonancia nuclear magnética
GABA: ácido gamaaminobutírico
Introducción
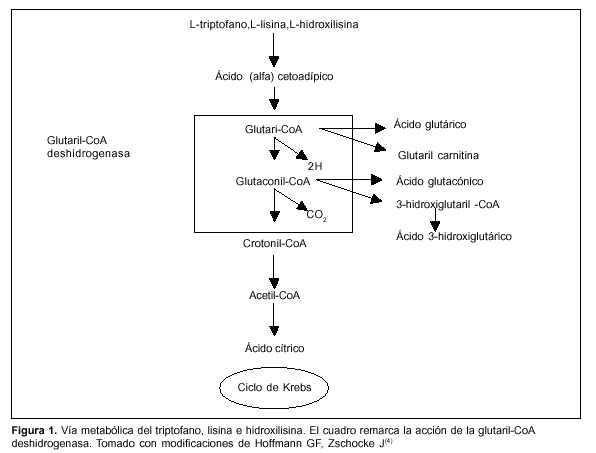
La aciduria glutárica tipo I (AGI) es un error congénito del metabolismo de los aminoácidos esenciales lisina y triptofano determinado por la deficiencia de la enzima mitocondrial glutaril-coenzima A deshidrogenasa (GD)(1). Fue descrita por primera vez en 1975 por Goodman y colaboradores(2) y se caracteriza clínicamente por distonía, discinecia, macrocefalia, cuya lesión orgánica está caracterizada por degeneración estriatal, en particular de los núcleos caudado y putamen(3). Como consecuencia de la deficiencia enzimática hay aumento de ácido glutárico, 3-hidroxiglutárico y glutacónico, que son los marcadores bioquímicos detectados en orina(1,3) (figura 1)(4).
]]> El tratamiento con L-carnitina y una rápida intervención ante episodios hipercatabólicos con líquidos, electrolitos y glucosa previene los síntomas neurológicos en pacientes sin daño estriatal al momento del diagnóstico y evita mayor daño en los pacientes sintomáticos(3,5,6). El efecto del tratamiento con riboflavina y restricción dietaria de aminoácidos glutarigénicos es menos clara(3). El mecanismo de herencia es autosómico recesivo. La enfermedad es causada por mutación del gen responsable de codificar la enzima GD que se encuentra localizado en el cromosoma 19p13.2(7,8). Más de 70 mutaciones causantes de la enfermedad han sido identificadas(5,8-10). La prevalencia a nivel mundial se estima en 1/40.000 recién nacidos caucásicos, si bien sería variable para diferentes poblaciones(7,11).Se presenta este caso por ser una afección poco frecuente y que en su presentación aguda puede confundirse con una encefalitis y en la crónica con una parálisis cerebral distónica progresiva de otro origen.
Caso clínico
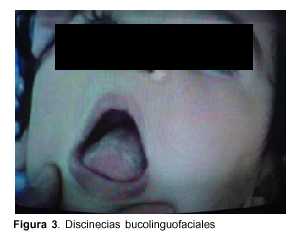
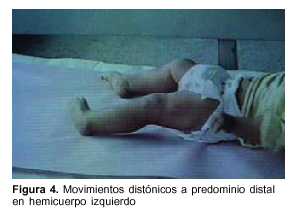
Lactante de 1 año, raza blanca, sexo femenino, con antecedentes familiares de padres consanguíneos (parientes tres grados aparte). Producto de primera gestación, embarazo normal, parto de término. Peso al nacer 4.500 g, longitud 53 cm, perímetro cefálico 35 cm. Test de Apgar 6/8. Buen crecimiento y desarrollo (figura 2). Comienza su enfermedad dos días después de una enfermedad diarreica aguda, caracterizada por trastorno de conciencia, crisis convulsiva generalizada tónica y movimientos anormales bucolinguofaciales y de las extremidades. El examen físico al ingreso mostró: perímetro cefálico 47 cm, peso 10.200 g, longitud 80 cm. Sin alteraciones morfológicas, vigil, hiporreactiva, movimientos anormales espontáneos caracterizados por discinecias bucolinguofaciales (figura 3) y movimientos distónicos a predominio distal en hemicuerpo izquierdo (figura 4). Hipotonía axial severa, sin sostén cefálico ni sedestación e hipertonía espástica, hiperreflexia, clonus y Babinski bilateral a predominio derecha.
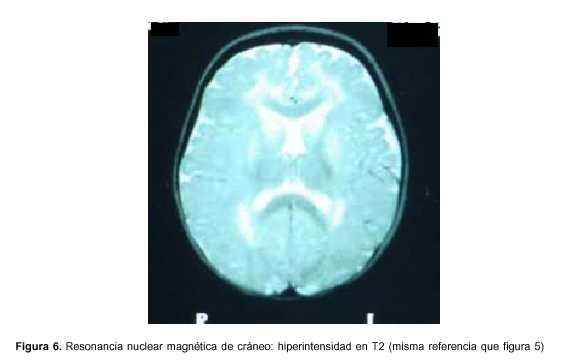
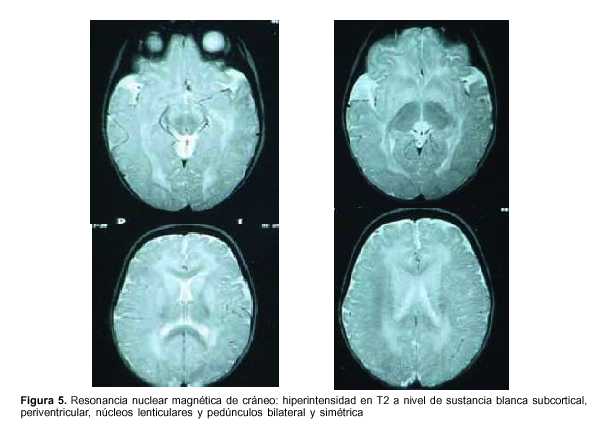
Exámenes paraclínicos realizados: hemograma: hemoglobina 9,8 g%, glóbulos blancos 11.900 elementos/mm3, plaquetas 377.000 elementos/mm3, azoemia 0,80 g/l, creatininemia 0,39 mg/dl, ionograma: Na 141mEq/l, K 4,3 mEq/l, Ca 1,2 mmol/l, glicemia 0,69 g/l, gasometría venosa: pH 7,36, exceso de base –6, HCO3 18,2 mEq/l, funcional y enzimograma hepático: bilirrubina total 0,24 mg/dl, bilirrubina directa 0,03 mg/dl, bilirrubina indirecta 0,21 mg/dl, transaminasas glutámico oxalacéticas (TGO) 123 U/l, transaminasas glutámico pirúvica (TGP) 101U/l, gamaglutamiltranspeptidasa 28 U/l, fosfatasa alcalina 479, lactato deshidrogenasa (LDH) 601, colinesterasa 9.183; examen de orina: límpida, vogel 2, ácida, densidad 1020, sin elementos anormales, líquido cefalorraquídeo (LCR): citoquímico incoloro, glucosa 0,54 g/l, proteínas 0,32, 10 elementos/mm3, a predominio mononucleares, estudio bacteriológico del LCR estéril, reacción en cadena de polimerasa en LCR para grupo herpes virus tipo 1 y 2 negativo, inmunoelectroforesis de LCR normal. Ácido láctico de LCR normal, HIV negativo, VDRL negativo, electroencefalograma (EEG): sufrimiento cerebral difuso sin actividad epileptógena focal, tomografía axial computarizada (TAC) de cráneo: normal, resonancia nuclear magnética (RNM) de cráneo: hiperintensidad en T2 a nivel de sustancia blanca subcortical, periventricular, núcleos lenticulares y pedúnculos bilateral y simétrica (figuras 5 y 6), ácidos orgánicos urinarios: aumento del ácido 3-hidroxiglutárico y ácido glutárico, carnitina plasmática: total: 32 mmol/l (valor normal 46-70); libre: 25 mmol/l (valor normal 41-58); libre %: 78 (valor normal 80%-90%).
Al ingreso con diagnósticos presuntivos de encefalitis viral o encefalomielitis difusa aguda se inicia tratamiento con anticonvulsivantes, antivirales (aciclovir) por 72 horas, corticoides (metilprednisolona) que se suspende al establecerse el diagnóstico de AGI. Se inicia entonces tratamiento específico con dieta hipercalórica, hipoproteica, riboflavina, vigabatrim y L-carnitina. Al alta se advierte al pediatra tratante sobre la importancia del tratamiento preventivo con glucosa intravenosa, líquidos y electrolitos ante eventuales estados hipercatabólicos a fin de evitar mayor daño neurológico.
Posteriormente no agrega sintomatología neurológica y a seis meses del inicio de su enfermedad se observa una detención del desarrollo psicomotor con estabilización de los síntomas, no habiendo reiterado crisis convulsivas.
]]>
Discusión
Las manifestaciones neurológicas son muy comunes en los trastornos de los ácidos orgánicos. Un grupo de estos desórdenes, dentro del cual se encuentra la AGI, llamados por algunos autores acidosis orgánicas "cerebrales", no se acompañan habitualmente de acidosis metabólica, cetosis, hipoglicemia, aumento de ácido láctico, como sucede en las acidosis orgánicas más conocidas(5).
Nuestra paciente se presenta con la forma clásica de AGI que se inicia entre los 2 y los 37 meses de edad con síntomas agudos(6). Consiste en la aparición brusca en el curso de una infección banal, inmunización, ayuno prolongado u otra situación hipercatabólica, de síntomas neurológicos que involucran principalmente el sistema extrapiramidal, siendo el desarrollo madurativo generalmente normal hasta ese momento(3,5,6). La paciente presentó, en forma brusca, hipotonía severa, movimientos distónicos principalmente bucolinguofaciales, irritabilidad, convulsiones y alteraciones piramidales sin acidosis metabólica. La enfermedad fue interpretada inicialmente como una encefalitis. Pasado el episodio agudo se produce una mejoría incompleta persistiendo la distonía y la coreoatetosis(12,13), si bien hay descripciones de algún caso con remisión completa luego del episodio agudo(6). No es infrecuente la disfunción hepática durante la descompensación(3), como fue detectado en nuestro caso con aumento de GOT y GPT. Otra forma de presentación aguda menos frecuente de la AGI es la llamada crisis metabólica con hipoglicemia hipocetósica y acidosis metabólica que progresa como si se tratase de un síndrome de Reye y que tendría como un importante factor causal deficiencia de carnitina (c). En la AGI se produce una deficiencia secundaria de carnitina, con relación esterificada/libre frecuentemente elevada(14). En nuestro caso, al momento de la evaluación, había un discreto descenso de carnitina total con leve disminución del porcentaje de la libre.
La forma crónica de la AGI es con retraso madurativo y progresiva instalación de distonía y coreoatetosis, siendo los pacientes catalogados muchas veces como una parálisis cerebral distónica(3,5,14,15). Esta afección ha sido diagnosticada en pacientes con el único signo clínico de macrocefalia desde el nacimiento o perímetro cefálico que aumenta más de lo esperado en los primeros meses de vida(15,16). Suele llamar la atención la discrepancia notoria, en algunos enfermos, entre el severo impedimento motor y las normales o casi normales funciones intelectuales(3,6,17).
Hay individuos con AGI catalogados como presintomáticos o sea que tienen la deficiencia enzimática pero no tienen síntomas. El diagnóstico en estos casos se efectúa cuando por su situación de alto riesgo son estudiados los hermanos sanos de un enfermo(6,18). Desde el punto de vista patogénico, si bien se ha visto en un paciente con AGI baja concentración de ácido gamaaminobutírico (GABA) en los núcleos caudado y putamen y se conoce la capacidad inhibitoria que tanto el ácido glutárico como el 3-hidroxiglutárico tienen sobre la enzima que lo sintetiza (ácido glutámico decarboxilasa), no se sabe con certeza si solamente la inhibición enzimática podría ser la causante del daño neuronal(3).
]]> Tanto los pacientes sintomáticos como los presintomáticos y aquellos con macrocefalia habitualmente tienen características neuroimagenológicas particulares: dilatación de la cisterna insular, regresión de los lóbulos temporales e hipodensidad de los núcleos lenticulares(19); puede verse también aumento de LCR a nivel de los lóbulos temporales y frontales así como compromiso de la sustancia blanca(3,19,20). Este aumento puede facilitar el sangrado de los vasos que atraviesan la zona ante un mínimo traumatismo, determinando sangrado subdural crónico y hematoma a veces acompañado de hemorragia retiniana. Estos hechos han llevado al diagnóstico de maltrato infantil en algunos casos(6). Nuestra paciente presenta compromiso de sustancia blanca y de los núcleos lenticulares bilaterales, coherente con los hechos clínicos.El diagnóstico se efectúa poniendo en evidencia la presencia de ácido 3-hidroxiglutárico, el cual es específico de la afección ya que no se ha encontrado en otras afecciones, a diferencia del ácido glutárico que dio el nombre original a la enfermedad pero que puede encontrarse aumentado en orina en otros errores congénitos del metabolismo y en situaciones adicionales como en la cetosis severa(3,21). El diagnóstico puede confirmarse con estudio de actividad de la enzima GD o estudios moleculares, o ambos. Nuestro caso presenta en orina, ácido glutárico y 3-hidroxiglutárico. Para algunos autores, si un paciente tiene elementos clínicos sugestivos de la afección y signos neurorradiológicos consistentes con el mismo, pero no se detecta el perfíl bioquímico a pesar de repetidas evaluaciones, debe efectuarse el estudio de la actividad de GD en los linfocitos o en fibroblastos cultivados a fin de confirmar o descartar el diagnóstico(3,5).
El tratamiento está dirigido a evitar el compromiso neurológico en los asintomáticos y a evitar mayor lesión en los pacientes sintomáticos(5,13,16,17). En todos los casos debe administrarse L-carnitina en dosis de 30 a 100 mg/kg/día dividida en tres tomas. Su aporte es fundamental en los primeros cinco años de vida, posteriormente puede usarse puntualmente ante la presencia de una enfermedad intercurrente dado que a esta edad las posibilidades de una crisis encefalopática son muy bajas. La dieta hipoproteica con restricción especial de lisina (60-100 mg/kg/día) y de triptofano (10-20 mg/kg/día), si bien se recomienda clásicamente, su eficacia es discutida pues no está probado que la misma prevenga las crisis encefalopáticas ni mejore las secuelas(3). A fin de intentar aumentar el contenido cerebral de GABA se administra vigabatrin, estando desaconsejado el uso de ácido valproico ya que éste aumenta la pérdida de carnitina. El baclofeno puede usarse para combatir la espasticidad, pero en algunos pacientes puede empeorar la hipotonía axial(1,16,22,23). Una rápida intervención ante episodios hipercatabólicos con líquidos, electrolitos y glucosa, incluso con insulina si fuera necesario, previene los síntomas neurológicos en pacientes sin daño estriatal y evita mayor daño en los pacientes sintomáticos(3,5,6,17,23). En nuestro caso, una vez establecido el diagnóstico de AGI, se inicia tratamiento específico con dieta hipercalórica, hipoproteica, riboflavina, vigabatrin y L-carnitina. Al ser la paciente del interior del país, al alta se advierte al pediatra tratante sobre la importancia de instituir el tratamiento preventivo en forma rápida ante eventuales estados hipercatabólicos a fin de evitar mayor daño neurológico.
Con respecto al pronóstico de esta enfermedad, una vez instalado el daño neurológico es irreversible, dependiendo la evolución de la prevención de las crisis encefalopáticas(13).
La frecuencia de la AGI en la población general oscila en valores que van de 1/30.000 a 1/40.000. En algunas comunidades cerradas es sensiblemente mayor, como entre los amish de Pennsylvania o entre los indios salteaux ojibway de Canadá(1,10). En nuestro país no existen datos al respecto.
Es una alteración hereditaria autosómica y recesiva. El asesoramiento genético para la pareja es de alto riesgo (1/4) de recurrencia. El gen humano que codifica la enzima GD está localizado en el cromosoma 19p13.2 extendiéndose a lo largo de 7 Kb y comprendiendo 11 exones y 10 intrones(8,24).
Conclusiones
Destacamos la importancia de pensar en esta enfermedad ante un niño de primera infancia con una encefalopatía aguda o progresiva con síntomas extrapiramidales así como en aquellos lactantes con macrocefalia y signos neurorradiológicos como los descritos (atrofia fronto-temporal, degeneración estriatal bilateral). Además, destacamos la importancia de investigar en los hermanos sanos del propósitus dado que el tratamiento precoz puede cambiar el curso de la enfermedad evitando el daño neurológico progresivo e irreversible.
]]>Agradecimientos
A la doctora Alma Martínez por su colaboración.
Summary
The first national clinical case of a child carrier a type I glutaric aciduria is reported on this paper. Type I glutaric aciduria is an inherited genetic disorder caused by a defect deshydrogenase mytocondrial glutaric-coenzime A, responsible for the metabolism of the amino acids lysine and tryptophan. The child presented with the classical form of the disease with neurologic symptoms involving the extrapiramidal system. 3-hydroxiglutaric acid defines diagnose, since it is specific to this affection. Treatment aims to prevent neurologic commitment in asymptomatic cases since brain damage is irreversible.
Résumé
]]>On présente le premier cas national d’un enfant porteur d’acidurie glutarique type I. Cette maladie est dûe à une erreur congénitale du métabolisme des aminoacides essentiels lysine et tryptophane, déterminé par disfonc-tionnement de l’enzyme mitochondriale glutaryl-Coenzyme A déshydrogénase. Chez notre patient, elle se présente de manière classique, avec des symptomes neurologiques aigus qui engagent surtout le système extrapyramidal. Le diagnostic se fait lorsqu’on constate la présence de l’acide 3-hydroglutarique, spécifique de cette maladie. Le traitement vise à éviter l’engagement neurologique dans les cas asymptomatiques, étant donné qu’une fois installé les lésions neuronales sont irréversibles.
Bibliografía
1. Prats Viñas J. Aciduria glutárica tipo 1: acidemia orgánica sin acidosis y con graves trastornos del movimiento. Neurología 2001; 16(8): 337-41.
2. Goodman SI, Markey SP, Moe PG, Miles BS, Teng CC. Glutaric aciduria: a "new" disorders of amino acid metabolism. Biochem Med 1975; 12(1): 12-21.
]]>3. Goodman S, Frerman F. Organic acidemias due to Defectos in Lysine Oxidation: 2-Ketoadipic Acidemia and Glutaric Acidemia. In: Scriver CR, Beaudet AL, Sly WS and Valle D, eds. The metabolic and molecular bases of inherited diseases. 8th ed. New York: McGraw-Hill, 2001: 2195-204.
4. Hoffmann GF, Zschocke J. Glutaric aciduria type I: from clinical, biochemical and molecular diversity to successful therapy. J Inherit Metab Dis 1999; 22(4): 381-91.
5. Hoffmann GF. Glutaric aciduria type l and related cerebral organic acid disorders. In: Fernandes J, Saudubray JM, Van der Berghe G, eds. Inborn metabolic diseases: diagnosis and treatment. Berlín: Springer-Verlag, 1996: 229-36.
6. Hoffmann GF, Athanassopoulos S, Burlina AB, Duran M, de Klerk JBC, Lehnert W, et al. Clinical course, early diagnosis, treatment and prevention of disease in glutaryl-CoA dehydrogenase deficiency. Neuropediatrics 1996; 27(3): 115-23.
7. Kölker S, Hoffmann GF. Glutaryl-CoA dehydrogenase deficiency. Orphanet encyclopedia, june 2001. http://www.orphanet.infobiogen.fr/data/patho/gb/uk-gdd.html. [consulta: ago 2003].
]]>8. Schwartz M, Christensen E, Superti-Furga A, Brandt NJ. The human glutaryl-CoA dehydrogenase gene: report of intronic sequences and of 13 novel mutations causing glutaric aciduria typeI. Hum Genet 1998; 102(4): 452-8.
9. Goodman SI, Stein DE, Schlesinger S, Christensen E, Schwartz M, Greenberg CR, et al. Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): review and report of thirty novel mutations. Hum Mutat 1998; 12(3): 141-4.
10. Ikeda H, Kimura T, Ikegami T, Kato M, Matsunaga A, Yokoyama S, et al. Novel mutations of the Glutaryl-CoA dehydrogenase gene in two japanese patients with Glutaric Aciduria type I. Am J Med Genet 1998; 80(4): 327-9.
11. Superti-Furga A, Hoffmann GF. Glutaric aciduria type 1 (glutaryl-CoA dehydrogenase deficiency): advances and unanswered questions. Report form an international meeting. Eur J Pediatr 1997; 156(11): 821-8.
12. Chamoles N. Encefalopatías evolutivas. Errores congénitos del metabolismo. In: Fejerman N, Fernández Álvarez E. Neurología pediátrica. 2ª ed. Buenos Aires: Médica Panamericana,1997: 326-7.
]]>13. Bjugstad KB, Goodman SI, Freed CR. Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type 1. J Pediatr 2000; 137(5): 681-6.
14. Merinero B, Pérez-Cerdá C, Font LM, García MJ, Aparicio M, Lorenzo G, et al. Variable clinical and biochemical presentation of seven Spanish cases with glutaryl-CoA-dehydrogenase deficiency. Neuropediatrics 1995; 26(5): 238-42.
15. Haworth JC, Booth FA, Chudley AE, deGroot GW, Dilling LA, Goodman SI, et al. Phenotypic variability in glutaric aciduria type I: report of fourteen cases in five Canadian Indian kindreds. J Pediatr 1991; 118(1): 52-8.
16. Hoffmann G, Trefz FK, Barth PG, Böhles HJ, Lehnert W, Christensen E, et al. Macrocephaly: an important indication for organic acid analysis. J Inherit Metab Dis 1991; 14(3): 329-32.
17. Aurebach VH. Errores innatos del metabolismo. In: Nelson WE. Tratado de Pediatría. 15º ed. Barcelona: Salvat, 1997: 509-16.
]]>18. Amir N, Elpeleg ON, Shalev RS, Christensen E. Glutaric aciduria type I: enzymatic and neuroradiologic investigations of two kindreds. J Pediatr 1989; 114(6): 983-9.
19. Mandel H, Braun J, el-Peleg O, Christensen E, Berant M. Glutaric aciduria type I: brain CT features and a diagnostic pitfall. Neuroradiology 1991; 33(1): 75-8.
20. Corral I, Martínez Castrillo JC, Martínez-Pardo M, Gimeno A. Aciduria glutárica tipo l: diagnóstico en el adulto y variabilidad fenotípica. Neurología 2001; 16(8): 377-80.
21. Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann GF. Sensitivity and apecificity of free and total glutaric acid and 3-hydroxyglutaric acid mesurements by stable-isotope dilution assays for diagnosis of glutaric aciduria type I. J Inherit Metab Dis 1999: 22(8): 867-82.
22. Castro-Gago M, Novo-Rodríguez M, Eiris-Puñal J. Tratamiento de las enfermedades mitocondriales durante la infancia y adolescencia. Rev Neurol 1998; 26(supl 1): S92-8.
]]>23. Ledesma F, Bay L, Halac A, Tenenbaum S, Acevedo E, Quintana S. Actitudes médicas en niños con errores congénitos del metabolismo y daño neurológico severo al momento del diagnóstico. Arch Argent Pediatr 2002; 100(2): 142-7.
24. Busquets C, Merinero B, Christensen E, Gelpí JL, Campistol J, Pineda M, et al. Glutaryl CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically and biochemically distinct. Pediatr Res 2000; 48(3): 315-22.
{kind=link}
{kind=link}