











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
1. Introduction
In Uruguay, strawberry crop is distributed in two main areas: Canelones and San Jose in the south, and Salto in the north. The value of this crop in farm systems lies in the intensive use of resources, workforce and capital, generating high profit margins per unit area1.
An important number of death of plants has affected strawberry crop in the north of the country associated with root and crown diseases caused by a complex of pathogens that emerged in 2015. This mortality reached losses between 30-50 % in crops destined for fruit production. In addition, the loss was total in mother plants for nurseries2. Within the complex, there were identified Neopetalotiopsis sp. (81.8 %), Fusarium spp. (75.7 %), Cylindrocarpon spp. (42.4 %), Rhizoctonia spp. (24.2 %), among other genres with lower frequencies3. The species identified in Uruguay are Colletotrichum fragariae, Verticillium albo-atrum, Rhizoctonia fragariae, Macrophomina phaseolina4, Phytophthora cactorum, V. dahliae5 and Neopestalotipsis clavispora6. In addition, Dactylonectria novozelandica and D. macrodidyma were associated with the death of strawberry plants, however they were not reported as pathogens in other regions3.
Pathogenicity and species of Cylindrocarpon are unknown in Uruguay. The aim of this work was to characterize isolations of Cylindrocarpon spp. by molecular and morphological analyses and test their pathogenicity in strawberry plants.
2. Materials and methods
2.1 Isolates of Cylindrocarpon spp.
Three isolates of Cylindrocarpon spp. were used from the collection of the Phytopathology Laboratory (School of Agronomy, Udelar). They were collected from naturally infected strawberry fruiting plants in 2018 in Salto (Table 1) and stored at −20 °C on filter paper inside sterilized envelopes. The isolates were randomly selected whilst still considering morphology, specifically the characteristics of the colony.
2.2 Molecular identification
Molecular identification was carried out using DNA extracted from three colonies of the fungi in potato dextrose agar (PDA) culture medium incubated for 13 days at 24 °C. The DNA concentration of the samples was determined using a Nanodrop 2000 Spectrophotometer Thermo Scientific, adjusting the concentration to 25 ng/μl. Genomic DNA was extracted using Quick-DNA™ Fungal/Bacterial Miniprep Kit (Zymo Research, USA). The amplification was performed by PCR with the primers CylH3F (AGGTCCACTGGTGGCAAG) and CylH3R (AGCTGGATGTCCTTGGACTG), of a partial region of Histone (H3) gene7. The cycling conditions were: initial denaturation at 94 °C for 2 minutes, followed by 35 cycles at 94 °C for 30 seconds, 60 °C for 30 seconds and 72 °C for 45 seconds, and a final extension of 72 °C for 10 minutes. PCR amplification was carried out in a PTC-100 Peltier Thermal Cycler. The master mix of the reaction was composed of 10X buffer (50 mMKcl, 10 mM Tris HCl (pH 8)) 2.5 μl, nucleotides (DNTP) 0.8 μM, 0.5μl of each of the primers, DNA 1μl, DMSO 0.5 μl, and Taq polymerase (MgCl included) 0.2 μl was added, the final reaction volume was 25 μl reached with 19 μl MQ water. Amplification products were analyzed by electrophoresis using 2 μl of sample and 1 μl of DYE loading buffer loaded on 1 % agarose gel, a molecular weight marker of 100 bp (Gene Ruler 1 kb DNA Ladder Plus, Fermentas, Germany) and a negative control without DNA were also added. PCR products were sequenced using the service of Macrogen Inc., Korea (www.macrogen.com). The obtained sequences were manually corrected using the program MEGA version 5.18, and aligned with similar sequences obtained from GenBank with BLAST analysis to identify the species used.
2.3 Morphological identification
For the morphological characterization, it was proceeded according to what is described by Halleen and others9. One pure isolate plated on PDA culture medium incubated for 20 days at 24 °C under ultraviolet (UV) light was used. Fungal colony was characterized by color and texture, using the description by Domsch and others10. Microscopic observations were performed on an Olympus CX41 optical microscope at 40× magnification, registering the length and width of 50 conidia through the program DinoCapture 2.0. In addition, characteristics of the conidia were observed such as its shape and number of septa.
2.4 Pathogenicity tests
Three fungi were incubated in Petri dishes with PDA under UV light. 25 days later, the medium containing conidia were macerated, passing it several times through sterilized distilled water (SDW) with Drigalski spatula. The conidial suspension resulted from the maceration of one plate diluted in 25 ml of SDW, the concentration of the inoculum was adjusted to 106 conidia/ml. Pathogenicity assays were performed in 20 strawberry plants “INIA Guapa” with 69 days old. Each isolate was inoculated in five plants; roots were dipped for 30 minutes with the inoculum and planted in pots of one liter with Bas Van Buuren substrate, covered with nylon bags for 24 hours. The plants were held at 20±5 °C and 80 % relative humidity in a growth chamber with a 14-hour photoperiod. Five strawberry pots were used as control, with the same treatment, except that the roots were dipped into SDW without conidia. The plants were placed randomly and periodically watered with distilled water.
2.5 Re-isolations from symptomatic plants
Once the lesions developed, fungi were re-isolated from the inoculated plants. Symptomatic plants were washed under running water to remove the soil. Then five pieces from infected tissues from root and crown were cut and superficially disinfected with 70° alcohol for 1 minute, rinsed three times with SDW, and air-dried in a laminar flow hood. They were first plated on water-agar with 0.2 g of streptomycin sulfate, after 72-96 hours at 24 °C they were spiked onto PDA and incubated for 20 days. The re-isolated fungi were identified by DNA extraction and subsequent PCR amplification and sequencing.
3. Results
3.1 Molecular identification
BLAST analysis of three isolates deposited in GenBank (accession number: MT892668, MT892669 and MT892670, sequences of 33Cyl, 92Cyl and 128Cyl, respectively) showed 100 % identity with D. novozelandica (MK409914.1) for partial his gene.
3.2 Morphological identification
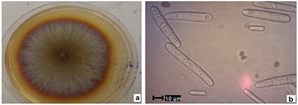
After 20 days, fungal colonies were orange to dark-brown and cottony aerial mycelium (Figure 1a). The fungus produced many conidia. Under a microscope, hyaline conidia with smooth, mostly straight walls with rounded ends (Figure 1b) of variable size between 27.3-45.8×4.1-7.3 μm were observed, presenting mostly 3 septa, coinciding with data reported11.
3.3 Pathogenicity tests and re-isolations
Nine days after inoculation, all plants showed necrosis in the outer leaves (Figure 2). 137 days after inoculation, necrosis was observed in the root systems and incipient crown lesions (Figure 3). From the symptomatic plants, D. novozelandica was consistently re-isolated from symptomatic tissues in crown and root, while the plants without inoculation remained healthy and no fungus was recovered from their tissues.


4. Discussion
Dactylonectria novozelandica has been reported as a vine pathogen in regions of America, Europe, and Oceania11. It was further reported as an avocado pathogen in Australia12. Although it was not reported as a strawberry pathogen, D. novozelandica was associated with the mortality of strawberry plants in the north of Uruguay. Previously in this region, Cylindrocarpon spp. (Dactylonectria spp.) was isolated from symptomatic plants with a high frequency -ranking third in terms of frequency3.
In this work D. novozelandica is causing root and crown rot in strawberry as it was expected according to the background in Uruguay.
5. Conclusion
Dactylonectria novozelandica was identified molecularly and morphologically, and then its pathogenicity was tested. According to this, the present work is the first to determine the pathogenicity of D. novozelandica in strawberry. Therefore, this species is part of the pathogens complex that causes root and crown rot in Uruguayan strawberries.
This is the first report of D. novozelandica causing root and crown necrosis on strawberry in Uruguay.

