











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Introducción
La vasculitis por IgA (antes denominada Purpura de Schönlein-Henoch) está clasificada como una vasculitis de pequeño vaso, mediada por depósito de inmunocomplejos (IC) que contienen IgA. 1,2,3,4,5,6
Es la vasculitis más frecuente en la infancia, con una proporción niños - adultos de 150-200 a 1. 3 En el adulto, se puede observar en todas las edades, siendo la edad media de presentación los 50 años (rango 15 -86 años). (2,6,7
La incidencia anual en España es de 11/100.000 personas en menores de 14 años, y de 1,5-14 casos por millón de habitantes en la edad adulta. 2
La clínica y el pronóstico de la vasculitis IgA del adulto, difiere de la de los niños. Mientras que en éstos últimos no hay diferencia entre sexos 8, en el adulto predomina en el sexo masculino (ratio hombre/mujer: 1,7). 6 En la infancia el curso suele ser benigno y autolimitado (2,4,9; mientras que en adultos suele presentarse como una vasculitis persistente, con recaídas frecuentes (30%), con mayor riesgo de purpura severa, es más común la nefropatía y la progresión a insuficiencia renal (13%) y enfermedad renal terminal (11%). 2,4,5,6,7,8
Habitualmente se presenta como una vasculitis sistémica, pero puede estar limitada a un solo órgano (ejemplo vasculitis IgA limitada a la piel). 3,4
Para algunos autores, la nefropatía a IgA representaría una variante de la vasculitis IgA, limitada a los riñones 3 mientras que para otros, sería una entidad independiente.9 La nefropatía IgA comparte con la vasculitis IgA similitudes clínicas y patológicas; es difícil distinguir a la nefropatía IgA de una vasculitis IgA limitada al riñón. 4
Un pequeño subgrupo de vasculitis IgA en adultos, está relacionado a malignidad, particularmente el cáncer de pulmón. 6,7
Es clásica, la presentación clínica con la tríada de purpura palpable, artralgias o artritis y dolor abdominal 2,3,4,5 y puede presentar glomerulonefritis (GN) con depósitos mesangiales de IgA. La afectación renal y el compromiso gastrointestinal son las principales causas de morbimortalidad en adultos. 3,4,5
Su patogenia es desconocida, se han implicado factores ambientales y genéticos, anomalías de la inmunidad innata y adquirida, teniendo un rol principal el depósito de complejos inmunes que contienen IgA1 deficiente en galactosa (Gd-IgA1) en la pared vascular. 4,5 La inmunidad de las mucosas, especialmente los órganos linfoides gastrointestinales, jugarían un papel clave en la patogénesis de la vasculitis IgA. La producción de Gd-IgA1 y autoanticuerpos contra Gd-IgA1, determinaría la formación de IC que se depositarían en los tejidos activando el proceso inflamatorio que genera el daño tisular. 4,10
A continuación se reporta el caso de un paciente con diagnóstico de vasculitis IgA del adulto.
Caso clínico
Hombre de 60 años con antecedentes personales de ex fumador y ulcus gastroduodenal perforado 20 años previos. En 2018 presentó prueba de sangre oculta en heces (PSI) positiva con endoscopias normales. Antecedentes familiares de un hermano con trasplante renal por insuficiencia renal crónica (IRC) secundaria a nefropatía IgA.
Comenzó 6 meses previos al ingreso, de forma súbita, con episodios recurrentes de petequias cutáneas, palpables, no pruriginosas ni dolorosas, sin compromiso mucoso, a predominio de miembros inferiores que llegaban hasta 1/3 distal del abdomen. Concomitantemente, artralgias de codos y rodillas que luego no reitera. No ingesta de fármacos, ni clínica infecciosa precediendo el inicio del cuadro.
Consultó en servicio de urgencias, en ese momento la analítica era normal. Se indicó Prednisona (PDN) en pauta decreciente sin mejoría.
A los 3 meses del inicio del cuadro había instalado proteinuria y hematuria, con deterioro de la función renal y elevación de los reactantes de fase aguda, leucocitosis con neutrofilia y anemia leve.
Dado el rápido deterioro de la función renal con sedimento urinario activo, ingresó para realización de punción biopsica renal (PBR) diagnóstica, completar valoración y definir conducta terapéutica. Del examen físico al ingreso se a destacaba buen estado general, en apirexia. Normotenso. A nivel de miembros inferiores la presencia de petequias en fase de resolución. Resto del examen normal.
Se realizaron los siguientes estudios para evaluación etiológica.
Anticuerpos antinucleares (ANA): positivos 1/160 patrón nucleolar
Anticuerpos anti-citoplasma de neutrófilo (ANCA), antifosfolípido (AAF), anticuerpos anti membrana basal glomerular, crioglobulinas, Factor Reumatoideo (FR) y anticuerpos anti C1q: negativos
Complementemia: normal
Inmunoglobulinas séricas: IgA elevada 3,75 g/l. IgG e IgM normales.
Serología para virus hepatitis B (VHB), virus hepatitis C (VHC), virus inmunodeficiencia humana (VIH): negativos
Radiografía de tórax, electrocardiograma, ecografía aparato urinario: normales.
PSI: negativo.
No se realizó biopsia cutánea por tratarse de petequias evolucionadas.
Se realizó tratamiento con Enalapril 2,5 mg vo/día y PDN a dosis de 1mg/kg/día, en espera del resultado de la PBR para conducta terapéutica definitiva.
Al momento del alta, la función renal era estable con leve mejoría, disminución de los reactantes de fase aguda y de la leucocitosis. Durante la internación presento anemia macrocítica y linfopenia leves.
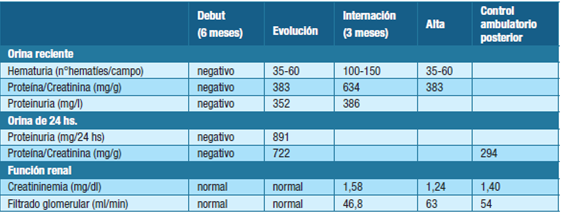
En la tabla 1 se presenta la evolución de la analítica del paciente.
Resultado de la PBR. Histopatológico (HP): ausencia de proliferación endocapilar o extracapilar, de necrosis o esclerosis segmentaria. Fibrosis intersticial y atrofia tubular afectando 30% del parénquima cortical. Inmunofluorescencia directa (IFD): depósitos glomerulares mesangiales para IgA +++/+++, C3 ++, IgM ++. Depósitos mesangiales de cadenas ligeras de predominio lambda+++ kappa +/-.
Discusión
El paciente presentó petequias, que por ser palpables, con una distribución cutánea característica (predominio en miembros inferiores) y no acompañarse de compromiso mucoso, constituye un síndrome purpúrico petequial de causa vascular (no plaquetopénico), destacando los episodios recurrentes. Aunque no se realizó biopsia cutánea debido a lo evolucionado de las petequias, desde el punto de vista HP, la causa más común del purpura vascular, es la vasculitis leucocitoclástica cutánea (VLC), no pudiéndose avanzar más en los diagnósticos clínicos en este punto, ya que varias entidades pueden causar la misma.
Presentó una insuficiencia renal aguda (IRA), ya que en la analítica inicial, la función renal era normal, y no existían alteraciones renales en la ecografía renal. El sedimento urinario activo en este contexto clínico junto a la elevación de los reactantes fase aguda, orientaba a la nefropatía de causa glomerular e inflamatoria, destacando la rápida progresión del cuadro. Todo esto en un varón de 60 años, con antecedentes familiares de nefropatía IgA (lo que orientaba, a una predisposición genética a enfermedad autoinmune mediada por IC) y con IgA sérica elevada.
Se realizó diagnóstico clínico presuntivo de vasculitis sistémica activa, con sospecha diagnóstica de vasculitis por IgA del adulto.
Los diagnósticos diferenciales planteados fueron las vasculitis asociadas a ANCA (VAA), sobre todo con la Poliangeítis Microscópica y la Granulomatosis con poliangeítis (GPA/Wegener). La ausencia de fiebre y repercusión general, así como de afección otorrinolaringológica (en el caso de la GPA) o compromiso pulmonar y/o neuropático (en ambas entidades), alejaban estos planteos diagnósticos. Además los ANCA fueron negativos. No era planteable la Granulomatosis Eosinofílica con poliangeítis (Churg- Strauss) dada la ausencia de asma, eosinofilia periférica y de afección pulmonar.
Otros diagnósticos diferenciales considerados fueron las vasculitis crioglobulinémica y la urticariforme hipocomplementemica (VUH) (también vasculitis sistémicas de pequeño vaso mediadas por depósito de IC).
En el caso de la vasculitis crioglobulinemica, no habían manifestaciones isquémicas con la exposición al frio, ni clínica de hiperviscosidad, así como tampoco evidencia de entidades que pudieran causarla, como las neoplasias hematoncológicas, tumores sólidos o las enfermedades autoinmunes sistémicas clásicas (EAS); las serologías virales (VHC), la determinación de crioglobulinas así como el FR fueron negativos, y la complementemia fue normal, todo lo cual, alejaba este diagnóstico.
Tampoco presentaba las típicas lesiones cutáneas urticariformes, tipo habón, de más de 24 hs de duración de la VUH (que es la que suele dar nefropatía); la complementemia fue normal y los AC anti C1q fueron negativos.
Las vasculitis secundarias a conectivopatías (Lupus Eritematoso Sistémico, Sjögren), a otras enfermedades reumatológicas (Artritis Reaumatoidea) o a neoplasias, no eran
planteables por el cuadro clínico y los resultados de los estudios.
No hubo exposición a tóxicos o ingesta de fármacos, ni infección clínicamente evidente previa al inicio del cuadro que pudieran explicar el mismo; y el compromiso sistémico, alejaba el diagnóstico de nefropatía primaria por IgA.
El paciente cumplía con los criterios actualmente disponibles para el diagnóstico de vasculitis IgA (aunque no universalmente aceptados): los de la clasificación para Purpura de Schönlein-Henoch propuestos por el Colegio Americano de Reumatología en 1990 11 y con los criterios diagnósticos de la Sociedad Europea de Reumatología pediátrica y la Liga Europea contra el Reumatismo (EULAR/PRINTO/PReS). 12
La vasculitis IgA tiene dos características principales, a saber:
1) A nivel HP la presencia de vasculitis leucocitoclástica: infiltración de la pared de pequeños vasos por polimorfonuclerares neutrófilos, con el fenómeno de leucocitoclasia (degranulación y fragmentación del núcleo de los neutrófilos o “polvillo nuclear”). También se observa edema endotelial, necrosis de la pared vascular, necrosis fibrinoide y extravasación de hematíes. 13
2) La IFD muestra depósitos de complejos inmunes con contenido predominante de IgA1 en la pared vascular (capilares, vénulas, arteriolas, o en el glomérulo renal). Estos depósitos pueden evidenciarse a nivel cutáneo y/o renal. 4,5
En este caso, el diagnóstico se confirmó por la presencia a nivel renal de depósitos glomerulares mesangiales con predominio de IgA en la IFD.
Una vez diagnosticada la EAS, se realizó un balance lesional, lo cual tiene implicancia pronóstica y terapéutica. En este paciente, la afección más trascendente era la renal, que si bien no constituía una IRA grave, o una proteinuria severa, preocupaba su instalación aguda y su rápida progresión.
La historia natural de la nefropatía en la vasculitis IgA (nefropatía-vasculitis IgA) en adultos, es menos conocida que en pediatría. 6 Existen discrepancias en cuanto a su frecuencia y la incidencia varía de 45 a 85%, según la definición de afectación renal que se utilice. 5,6,7,14
En adultos, no existe correlación entre la presentación inicial y el resultado renal a largo plazo, con posibilidad de remisión espontanea o la evolución a enfermedad renal terminal. 15 La nefropatía y el riesgo de progresión a insuficiencia renal en adultos es de aproximadamente 30% (0 a 50%) y el pronóstico es relativamente pobre, comparado con el de los niños. 5,6 Puede presentarse como una microhematuria y proteinuria leves, como síndrome nefrítico o nefrótico y/o insuficiencia renal y es indistinguible de la nefropatía IgA. 3,4,9,16
No existen guías que indiquen cuando realizar la PBR en adultos. Habitualmente se realiza ante la progresión de la nefropatía, con peoría de la proteinuria (> 500 mg/24 hs) o de la función renal (creatinina > 1,5 mg/dl), como sucedió en este paciente. 5,6,9
Los tipos HP de compromiso glomerular que pueden hallarse en la vasculitis IgA han sido clasificados por Pillebout et al, 6) y tienen connotación pronóstica y terapéutica.
Otra de las manifestaciones clínicas más frecuentes en la vasculitis IgA son las cutáneas, como lo fue en el caso clínico analizado.
El purpura palpable localizado en miembros inferiores, está presente en el 100% de los casos. El compromiso de miembros superiores y tronco es del 31,9% 5 y un 5% se localiza en cara o mucosas. 5,6,7,14 La duración media de las lesiones es de 10 días (6-15 días) y las recaídas son comunes (48% de los casos). 5,6 Las ulceras cutáneas, lesiones ampollosas hemorrágicas y el purpura necrótico (más común en mayores de 60 años), son infrecuentes. 6
A nivel cutáneo se produce una VLC, cuya expresión clínica característica es el purpura palpable (nódulo inflamatorio). Como la VLC puede presentarse en múltiples entidades, la evidencia del depósito de IgA por IFD en la biopsia cutánea (realizada por sacabocados, tipo punch) confirma el diagnóstico de vasculitis IgA.
Los depósitos de IgA desaparecen rápidamente y el infiltrado polimorfonuclear neutrófilo es sustituido por infiltrado mononuclear, por lo cual la biopsia cutánea debe realizarse en las primeras 24 a 48 hs desde el inicio de las lesiones, para un rendimiento diagnóstico óptimo. 2 En la práctica, a menudo se evita, sobre todo en niños con casos leves y con cuadro clínico característico. 5,9 La ausencia de tinción de IgA en la biopsia cutánea no excluye el diagnostico de vasculitis IgA. 9
Pueden presentarse artralgias (63,1%), artritis (37,4%) y mialgias, siendo lo más común, la oligoartritis no erosiva, de rodillas y tobillos. 5,6 En el caso analizado el compromiso articular fue leve (artralgias sin artritis).
La anemia en pacientes con vasculitis IgA, puede deberse a un sangrado clínico oculto, por isquemia/inflamación GI. En este caso, la anemia se interpretó como de causa multifactorial, ya que no presentó manifestaciones comunes de compromiso GI (64,5%) como dolor abdominal (64,5%), nauseas/vómitos (14,4%) ni diarrea sanguinolenta, melenas o rectorragia (12,9%) y el PSI fue negativo. 5 Se han reportado sangrados con requerimiento de transfusión, cirugía y como causa de muerte (11%). 6
Los niveles de IgA sérica fueron elevados, lo que es más común en pacientes con nefropatía, como en este caso, comparados con aquellos sin afección renal (41,8% vs 10%, respectivamente). 5 Sin embargo, los niveles séricos normales de IgA, no excluyen el diagnóstico de vasculitis IgA; su incremento varía entre 31-60%. 5,6,14
Aunque en la literatura se menciona que en la vasculitis IgA, es característica la negatividad para ANCA, ANA, FR, crioglubulinas, y que la complementemia es normal 2 otras series han reportado su positividad, incluidos los ANA (14,2 %) así como hiperleucocitosis (36,7%) como en este caso. 5,6,14)
El tratamiento de la vasculitis IgA ha sido poco investigado en adultos y suele basarse en la severidad del cuadro clínico. En caso de afectación renal, el tratamiento dependerá del tipo de lesión glomerual, y de la severidad de la proteinuria, pudiendo utilizarse desde tratamiento nefroprotector/antiproteinurico con un inhibidor enzima convertasa (IECA) y/o antagonista receptor angiotensina II (ARA II) hasta corticoides (CE) y/o inmunosupresores (IS). 2,9
No existen estudios controlados randomizados que avalen su eficacia, siendo una extrapolación de los tratamientos usados en otras EAS o de los estudios pediátricos. 14,15) Los CE son efectivos para las artralgias y el dolor abdominal pero es controvertido su beneficio en la afección renal y en la prevención de la enfermedad renal terminal. 14) Cuando se comparó la monoterapia con CE vs la terapia combinada de CE más Ciclofosfamida (CF) en formas severas de vasculitis IgA, no se hallaron diferencias significativas en cuanto a remisión de la enfermedad, resultado renal, muerte y eventos adversos, en 12 meses de seguimiento. Por lo tanto, el uso de CF en la vasculitis IgA también es controvertido. El Micofenolato de Mofetilo es útil como inductor de remisión y ahorrador de CE en la nefritis severa pero el efecto sobre la función renal a largo plazo no se conoce. 14).
Dado los potenciales efectos adversos de la CF, la monoterapia con CE sería una opción adecuada como terapia de primera línea excepto en la presentación severa, donde la decisión de usar CF (en pauta similar a la glomerulonefritis de las VAA), debe individualizarse, siendo su uso controversial 14,17 El tratamiento profiláctico con CE para prevención de la nefropatía-vasculitis IgA no está indicado. 9
En este paciente, en conjunto con nefrología se realizó tratamiento con un IECA, que ha demostrado su beneficio (o de los ARA II) en la proteinuria persistente y en las células mesangiales, previniendo y/o limitando la injuria glomerular secundaria. 9,18,19
Se mantuvo monoterapia con CE, supeditando el eventual agregado de un IS, a la evolución del paciente, particularmente a nivel renal.
Evolutivamente en el adulto, los rebrotes iniciales son frecuentes, con episodios más leves que el inicial 2,4,5.
En relación a su pronóstico, este paciente presentó IR inicial y proteinuria al momento de la PBR así como fibrosis intersticial y atrofia tubular del 30% (estos últimos elementos de cronicidad) todos los cuales constituyen factores de riesgo independientes para falla renal severa, 6,14 destacando la ausencia de proliferación endo o extracapilar, de necrosis o esclerosis segmentaria, que de estar presentes, también son factores de riesgo desfavorables para el pronóstico renal. 6,14)
En la serie de Pillebout et al 6 de adultos con vasculitis IgA, la mortalidad fue de 26%. Las causas de muerte fueron las neoplasias (27%), infecciones (16%), por el Purpura de Schönlein-Henoch el 11% (compromiso GI severo) y enfermedad cardiovascular 9%. En cuanto a la supervivencia renal, 11% desarrollaron ERT (48% a los 3 años) y el resto todos a los 10 años. 6 Veinticinco pacientes fueron dializados y a 12 se les realizó trasplante renal. Ninguno de los trasplantados, perdió su injerto por recaída. El 38% del total de pacientes, presentaron una insuficiencia renal significativa (13% severa, y 14% moderada). 6 Al final del seguimiento, la supervivencia fue del 74%. Solo 20% estaban en remisión clínica 6,18, el resto, 50% tenía microhematuria, 47% proteinuria mínima o moderada, y 8% tenía proteinuria en rango nefrótico. 6
Conclusiones
Se analizó el caso de un paciente que presentó un síndrome purpúrico petequial, alteración del sedimento urinario y una evolución rápida a la insuficiencia renal, de cuyo estudio etiológico surge el diagnóstico de vasculitis por IgA del adulto. Se destaca que la presentación clínica, evolución y pronóstico difiere de la de los niños y la importancia de la biopsia cutánea y/o renal con IFD y la alta sospecha clínica, para su diagnóstico, siendo el tratamiento controversial dado la falta de estudios controlados, randomizados que avalen su eficacia.

