











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Introducción
La amiloidosis refiere a un grupo diverso de enfermedades caracterizadas por el depósito extracelular de una proteína anómala e insoluble (amiloide), en los distintos tejidos de forma localizada o sistémica, determinando alteraciones funcionales y manifestaciones clínicas en los tejidos afectados. Pueden ser hereditarias o adquiridas. Su nombre proviene de la similitud con el almidón por su afinidad por los colorantes yodados. 1-4
Existen más de treinta proteínas que pueden producir depósitos de amiloide en los tejidos, dando lugar a diferentes afecciones como la enfermedad de Alzheimer o las enfermedades por priones. La forma más frecuente es la amiloidosis primaria o AL, que tiene una incidencia anual de 10 casos nuevos por millón de habitantes. Ésta se produce por acumulación de fragmentos de cadenas ligeras monoclonales (kappa o lambda), que sufren transformación conformacional anómala y se depositan como fibrillas de amiloide. La amiloidosis AL por cadenas lambda es más frecuente (75%) que por cadenas ligeras (25%) con un ratio k/l 1/3. 4-7
Otros tipos de amiloidosis incluyen; ATTR cuyo amiloide está formado por trastirretina mutante, la amiloidosis sistémica senil formada por trastirretina de tipo salvaje, amiloidosis AA (secundaria) formada por amiloide A sérico, entre otras. (1, 3)
La presentación clínica suele ser heterogénea, lo que determina el diagnóstico tardío. Las manifestaciones se correlacionan con la proteína involucrada, su variable afinidad por diversos tejidos y la cantidad de amiloide depositado. En la amiloidosis AL existen signos característicos pero poco frecuentes, presentes en el 15% de los pacientes.
Estos son la artropatía, el signo de la hombrera, la pseudohipertrofia muscular, el engrosamiento submandibular y el púrpura facial. La macroglosia se ve en el 10% de los pacientes, pudiendo producir obstrucción aérea, dificultad en la masticación y deglución, además de apnea del sueño. (1 - 4
Los órganos comprometidos más frecuentemente en la amiloidosis AL son corazón, riñón, sistema nervioso periférico, hígado e intestino. El riñón es el órgano más afectado, presentándose en la mayoría de los pacientes como síndrome nefrótico. El compromiso cardiaco es el segundo más frecuente, pero es el factor pronóstico más importante en cuanto a sobrevida. Este se manifiesta como insuficiencia cardiaca desde el inicio de la afección en el 20% de los pacientes con amiloidosis AL, existiendo también cardiomegalia, arritmias, deterioro de la función sistólica y diastólica, con un patrón de cardiopatía restrictiva. Más del 20% de los pacientes presentan síntomas de neuropatía periférica al diagnóstico (principalmente parestesias). La afectación suele ser simétrica, con compromiso de miembros inferiores y puede ser dolorosa. El síndrome del túnel carpiano es una manifestación común. La neuropatía autonómica (presente en el 65% de los pacientes) se presenta habitualmente como hipotensión ortostática, impotencia y disturbios gastrointestinales. La astenia y adelgazamiento son síntomas frecuentes al inicio, pero inespecíficos. 1-6
La confirmación diagnóstica de amiloidosis AL requiere de la presencia de síntomas característicos o síndromes asociados en ausencia de otra causa que las explique, la realización de biopsia tisular de un órgano involucrado con tinción rojo Congo positivo, sumado a la evidencia de que el depósito de amiloide esté formado por cadenas livianas y la demostración de un desorden monoclonal. 1,3
El rendimiento diagnóstico de la biopsia por aspirado de grasa subcutánea es del 84-88%, de la biopsia rectal 75-85%, de la médula ósea 50%, de glándulas salivales del 58%, biopsia renal y hepática del 94% y 97% respectivamente, alcanzando un rendimiento del 100% en la biopsia cardíaca. 1,3,7,8
El tratamiento y pronóstico varía según el tipo de amiloidosis, dependiendo entonces de la correcta identificación de la proteína involucrada. La determinación de la misma se realiza con inmunohistoquímica para cadenas livianas y AA, estudios genéticos para evaluar las mutaciones de la transtirretina (en caso de sospecha, no siempre es necesario realizarlo) y espectrometría de masa. Si bien esta última no está presente en nuestro país, es considerada el gold-estandar para la tipificación de la sustancia amiloide involucrada en todos los subtipos de amiloidosis y en la mayoría de los tejidos afectados. 1,2,3,7
Para la demostración del desorden monoclonal debe realizarse la evaluación inmunoproteica mediante proteinograma electroforético (PEF), inmunofijación (IFE), dosificación de inmunoglobulinas y cadenas livianas libres (FLC) tanto en sangre como en orina. También debe valorarse la infiltración medular mediante aspirado con inmunofenotipo, citogenético, FISH y biopsia. Es esencial realizar IFE tanto para determinar el tipo de inmunoglobulina, como en los casos en que la electroforesis es negativa, dada la mayor sensibilidad de este método. La detección de FLC es útil para el diagnóstico y monitorización de la respuesta al tratamiento. Esta además presenta mayor sensibilidad que el IFE (10 veces superior). En el 90% de los pacientes con amiloidosis AL se identifica la proteína monoclonal presente en suero u orina con IFE o PEF y su valor es característicamente bajo (menor de 20 g/l en más del 70%). 1-5
Confirmada la amiloidosis hay que proceder a la valoración de los órganos afectados. La evaluación cardiaca requiere realización de electrocardiograma (ECG), ecocardiograma y de ser posible resonancia nuclear magnética (RMN) con gadolinio (es el método más sensible y especifico, 80 y 94% respectivamente). Esta última presenta un patrón característico con realce subendocárdico con el contraste. Los biomarcadores cardíacos (proBNP y troponinas) son de gran utilidad para determinar el pronóstico de la enfermedad y son usados para estadificación. En casos de dudas diagnosticas el centellograma miocárdico es de gran utilidad. La valoración del compromiso renal incluye la detección de albuminuria, la identificación de proteinuria monoclonal y su tipificación. 1,3
El tratamiento depende del subtipo de amiloidosis; debiendo ajustarse a la edad, disfunción orgánica y la toxicidad en cada régimen, siendo guiados por los biomarcadores de respuesta hematológica y cardíaca, de existir este compromiso. En la amiloidosis AL sin tratamiento existe una sobrevida de 6 a 12 meses, la cual se reduce a 5 meses si existe compromiso cardíaco severo. 3,4,8
El tratamiento tiene como objetivo suprimir la síntesis de FLC, favorecer la reabsorción del amiloide optimizando así la función de los órganos afectados y mejorar la sobrevida. La quimioterapia se basa en regímenes desarrollados para mieloma múltiple. Existen escasos estudios randomizados prospectivos realizados en amiloidosis AL, por lo que la mayoría de la información surge de estudios retrospectivos. La introducción del trasplante autólogo de progenitores hematopoyéticos como parte del tratamiento ha representado un avance importante en esta enfermedad, sin embargo la mayoría no será candidato al mismo, tanto por la edad como por el compromiso de órgano. También se asocia con una alta mortalidad relacionada al trasplante (25%) por lo que sólo debe indicarse en casos seleccionados. 3,5,8-10
Si bien históricamente la combinación de Melfalán-Dexametasona oral ha sido el tratamiento de primera línea alcanzando sobrevidas de 60 meses, hoy regímenes basados en el inhibidor del proteosoma, Bortezimb son la piedra angular, logrando remisiones completas cercanas al 50%. Otros planes de segunda línea son los basados en inmunomoduladores como Lenalidomida y Pomalidomida. Nuevas drogas como Daratumumab muestran resultados prometedores. 8-10
El tratamiento se controla con FLC en sangre y orina y PEF. Las cadenas ligeras libres en suero tienen una vida media de horas en comparación a las semanas de las inmunoglobulinas intactas, por lo que son un indicador precoz de la respuesta al tratamiento. La respuesta hematológica se asocia directamente con la orgánica, lo cual es un factor pronóstico. 3
El objetivo del presente artículo es comentar un caso clínico y revisar la literatura.
Caso clínico
Paciente de sexo femenino, 68 años. Antecedentes personales de tumor carcinoide de apéndice diagnosticado en el año 1995. Tratamiento quirúrgico, sin tratamiento oncoespecífico. Seguimiento posterior, sin recidiva tumoral.
Historia de más de 15 años evolución de crecimiento excesivo y progresivo de músculos de cara, cuello, tronco, a predominio de músculos paravertebrales y de los cuatro miembros, de manera bilateral y simétrica, por lo que adquirió el aspecto físico de “levantador de pesas”. Negaba ingesta de tóxicos, hormonas o realización de ejercicio físico. En la evolución agregó debilidad y claudicación muscular, rigidez con importante limitación en la movilidad, sobre todo de las manos. Dolor generalizado a predominio de articulaciones de manos, sin elementos fluxivos. Disfagia progresiva para sólidos. Disfonía. Edemas de MMII bilaterales y simétricos a predominio perimaleolares.
Presento múltiples consultas a lo largo de los años con distintos planteos diagnósticos, enfocados a patología autoinmune (serología inmunológica negativas) se inició tratamiento con corticoides, hidroxicloroquina y azatioprina sin mejoría sintomática, por lo que se suspendieron, a excepción de los corticoides dado la peoría sintomática cuando se descendieron los mismos. Biopsia muscular en 2015 informada como “lipodistrofia muscular” y en 2016 “sin alteraciones en fibras musculares”. Punción de grasa subcutánea con tinción con rojo Congo no mostro depósitos de amiloide.
Al examen físico lúcida se constataba una paciente de aspecto atlético, con disminución de la bola de Bichat. Engrosamiento submandibular. Aumento difuso y simétrico de masas musculares de miembros superiores, con franco predominio de cintura escapular (signo de la hombrera) y pélvica y músculos paraespinales (Figuras 1 y Figura 2).
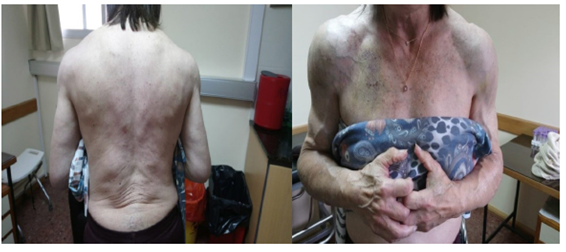
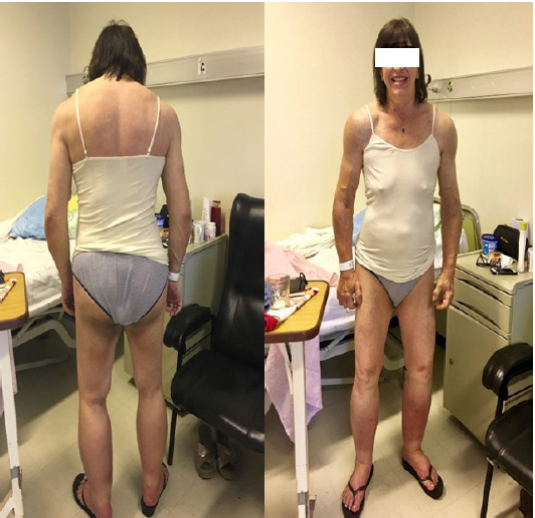
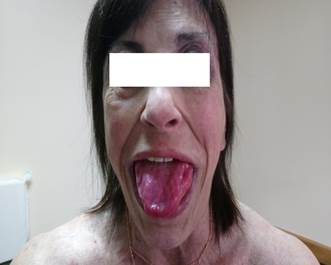
Las masas musculares eran duras “como madera” a la palpación, con desarrollo venoso marcado. La piel y mucosas estaban normocoloreadas y no presentaba lesiones. Se destacaba macroglosia con impronta dentaria (Figura 3). El resto del examen físico no presentaba alteraciones a destacar.
Se realizó tomografía axial computarizada (TC) cráneo, cuello, tórax, abdomen y pelvis que fue normal, tanto de estructuras profundas como de músculos.
La RNM de miembros inferiores informó la presencia de un linfedema bilateral y simétrico a nivel perimaleolar hasta raíz de muslo. Leve edema muscular difuso de distribución bilateral y simétrica a nivel de ambos músculos bastos internos y externos, ambos crurales y ambos sartorios, sin evidencias de colecciones intramusculares. Sin evidencia de atrofia muscular. El estudio electomiográfico (EMG) informó un patrón de tipo miopático. El estudio de la deglución mostró alteraciones del tiempo deglutorio faríngeo y esofágico sin aspiración. Transito faríngeo- esofágico: disminución de la distensibilidad parietal del tercio inferior del esófago, sin imagen de estenosis, se observó reflujo gastro-esofágico espontaneo en tercio superior de esófago de lento aclaramiento.
En busca de causas de miopatía se realizó determinación de creatinin-kinasa (CK) fue de 122 U/L (valores normales mujeres:96-140 U/L). Se completó la valoración con factor reumatoideo, anticuerpos anti-nucleares (AAN), anticuerpos Anti- DNA y anticuerpos anti-núcleo citoplasmáticos (ANCA) que fueron negativos. La determinación de TSH fue normal. Los marcadores tumorales antígeno carcino-embrionario (CEA), antígeno cáncer (CA 15-3 y CA 19-9 y CA 125) fueron negativos. La serología para virus hepatitis B, virus hepatitis C y virus imnunodeficiencia humana fueron negativos.
En 2018 se reiteró la biopsia muscular de deltoides con microscopia óptica convencional que observó material congofilo a nivel de paredes arteriales y entre fibras musculares estriadas, observándose con luz polarizada que algunos muestraban birrefringencia “verde manzana” característico de los depósitos de amiloide.
Con tinción de PAS, dichos depósitos son PAS positivos. Se completa la tipificación realizando espectrometría de masa del sector rojo Congo positivo. (Figuras 4)
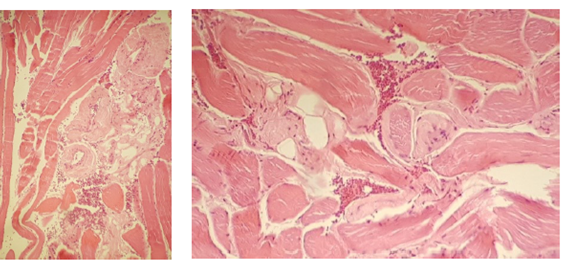
Figuras 4: Histología muscular. Se observa aumento del grosor y la hialinización de la pared del vaso y el espacio peri vascular dentro de las fibras musculares.
Con diagnóstico de amiloidosis AL con compromiso muscular por anatomía patológica se procedió a la identificación de clonalidad por lo que se realizó PEF e IFE en sangre que fue negativo. FLC en sangre: Kappa 198 mg/L (valores normales 3.3-19.4), Lambda 10.9 mg/L (valores normales 5.7-26.3), ratio κ/λ 18.17. Proteína de Bence Jones negativo. Mielograma con 6% plasmocitos, IF de médula ósea 1.36 % de plasmocitos siendo el 97.5% aberrantes clonales. El estudio citogenético de médula ósea fue normal. La biopsia de médula ósea (BMO) mostró elementos dishemopoyeticos. Población de células plasmáticas 5%, no se observó depósitos de amiloide en la muestra examinada.
Para la valoración de órganos afectados, a nivel cardiovascular se realizó ECG que fue normal, ecocardiograma transtorácico que mostró hipertrofia ventricular izquierda con fracción de eyección del ventrículo izquierdo (FEVI) conservada. La RNM cardíaca mostró aumento del espesor parietal del ventrículo izquierdo en los segmentos basales sin criterio de hipertrofia por masa. FEVI en el límite inferior de lo normal. Derrame pericárdico leve. En las secuencias de realce tardío no se evidencio fibrosis ni infiltración miocárdica. A nivel renal la determinación de azoemia y creatininemia era normal, no presentando proteinuria en el examen de orina.
Se realizó tratamiento en base a Ciclofosfamida, Bortezomib y Dexametasona días 1, 8, 15 y 22; a dosis reducida de dexametasona por la retención hídrica. Luego de tres ciclos, se evaluó la respuesta evidenciando una persistencia de la sintomatología, sin mejoría alguna y con valores de FLC iguales que al inicio.
En la evolución presentó progresión de su enfermedad, con aumento del dolor y debilidad muscular, falleciendo.
Discusión
La miopatía amiloide engloba miopatías clínica y etiológicamente heterogéneas, caracterizadas patológicamente por el depósito de amiloide en vasos sanguíneos intramusculares o en los elementos del tejido conectivo del músculo esquelético.
La acumulación de amiloide puede afectar a múltiples tejidos, incluido el músculo (miopatía asociada a amiloidosis sistémica), pero en ocasiones puede limitarse a los músculos esqueléticos (miopatía amiloide aislada). La hipertrofia muscular es debido al aumento del número y/o tamaño de las fibras musculares o por la infiltración de materiales no contráctiles. Característicamente es el síntoma de presentación de miopatías adquiridas o hereditarias como pueden ser la amiloidosis, hipotiroidismo, distrofias musculares, entre otras afecciones. 7,9-12
Se expone el caso de una paciente con miopatía amiloide a cadenas ligeras libres Kappa donde la hipertrofia muscular se presentó de manera uniforme y simétrica comprometiendo casi toda la musculatura, sin afectación de otros órganos.
Clínicamente se destacó la presencia de macroglosia, disfagia, signo de la hombrera, engrosamiento submandibular y consistencia muscular “tipo madera” características de la amiloidosis AL, tal como lo reportan distintos autores en la literatura.
Chapin et al. revisa una serie de casos obteniendo 79 pacientes con miopatía amiloidea, identificados por debilidad muscular y depósitos de amiloide en musculo esquelético al igual que la paciente del caso clínico citado. En concordancia, las características más frecuentes en estos fueron macroglosia y seudohipertrofia muscular, siendo menores los casos que se presentaron con debilidad muscular generalizada sin hipertrofia. (1,3,7,11-13
Liewluck y Milone exponen una revisión de casos, donde se encuentran 52 pacientes, incluidos 14 con miopatía amiloide aislada y 38 con amiloidosis sistémica. Se concluyó que, esta última, representa el subtipo más común de miopatía amiloide, siendo la amiloidosis AL la etiología más frecuente. 32 pacientes con amiloidosis AL tenían gammapatía monoclonal, como el caso de nuestra paciente. Igualmente el patrón de distribución de depósito de amiloide fue similar en ambos grupos, involucrando paredes de vasos intramusculares, perimisio o endomisio y algunas veces envolviendo fibras musculares. Casi el 70% de los pacientes con miopatía amiloide AL tenían miopatía como única manifestación inicial al inicio de la enfermedad. 12
La amiloidosis muscular es una entidad subdiagnosticada y debe ser considerada en el análisis diferencial de cualquier adulto con miopatía progresiva. El retraso en la confirmación etiológica es común, como en el caso expuesto por ser una forma atípica de amiloidosis AL; el diagnóstico suele ser temprano en las formas más frecuentes con compromiso multiorgánico.
Muchtar et al. demostraron que alrededor del 40% de los pacientes con miopatía asociada a amiloidosis AL fueron diagnosticados erróneamente a pesar de someterse a una biopsia muscular porque no se realizó tinción con rojo Congo, concordando con el caso expuesto donde se logra diagnostico con una tercer biopsia muscular. 1,3,7,9,14
El pronóstico es generalmente pobre con un tiempo medio hasta la muerte desde el inicio de los síntomas de 21.7 meses. Ocasionalmente, los pacientes tienen una supervivencia más larga. Se observó un curso más benigno, con una supervivencia a largo plazo en los pacientes con miopatía crónica, los cuales presentaban clínicamente distrofia muscular a predominio de cinturas escapular y pélvica, en los cuales la duración de los síntomas fue de 18 a 26 años antes del diagnóstico (más de 15 años en el caso expuesto). 7
Conclusiones
Se expone el caso de una paciente con miopatía amiloide a cadenas ligeras libres Kappa, afección generalmente subdiagnosticadas.
La amiloidosis es una causa rara de hipertrofia muscular, pero debe ser considerada en los diagnosticos diferenciales de miopatía en general, y en particular de las formas “hipertróficas”.
La presentación clínica presento macroglosia, disfagia, signo de la hombrera, engrosamiento submandibular y consistencia muscular “tipo madera” características de la amiloidosis AL. Al ser heterogénea determino el diagnóstico tardío, que se confirma con biopsia del tejido afectado, la demostración del depósito amiloide y la tipificación de la proteína que lo constituye. Si bien debe primar la sospecha diagnostica inicial, ya que aproximadamente el 40% de los pacientes con miopatía asociada a amiloidosis AL fueron diagnosticados erróneamente por biopsia como en el caso discutido.
En nuestro país disponemos de la mayoría de los estudios necesarios para el diagnóstico y estratificación pronóstica, así como para la evaluación del compromiso de órgano blanco. Aún está pendiente lograr la adecuada tipificación del amiloide, etapa crucial en la definición del tratamiento y pronóstico.
El tratamiento es individualizado, determinado por la edad, la disfunción de órganos y las toxicidades de los régimenes, debiendose guiar por biomarcadores y la respuesta cardiaca.

