











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
Las ganglionopatías también denominadas neuronopatías sensitivas, corresponden a un grupo de patologías con afección del soma de la primera neurona sensitiva del ganglio de la raíz dorsal y del ganglio de Gasser 1,2.
Las etiologías más habituales son adquiridas, entre ellas las paraneoplásicas (también denominada neuronopatía sensitiva carcinomatosa) que forman parte de los síndromes paraneoplásicos clásicos. La neoplasia más frecuentemente implicada es el cáncer broncopulmonar en su forma oat cell (hasta en el 80%) y los anticuerpos relacionados son el anti-Hu (en la mitad de los casos) 3,4. También hay formas hereditarias y degenerativas 1,3 - 5.
Se manifiestan por déficit sensitivo con arreflexia, eventualmente dolor y su distribución generalmente es difusa, de perfil no largo dependiente 1,6.
El patrón evolutivo es fundamental en la búsqueda etiológica y va desde la desaferentización total en 2 a 3 días hasta cuadros de evolución indolente durante décadas, definiéndose 3 patrones fundamentales, agudo, subagudo y crónico 1.
La forma aguda o neuronopatía sensitiva aguda alcanza su máxima expresión en 72 horas, las formas subagudas se instalan en semanas a pocos meses y las formas de evolución crónica se instalan en años 1.
En la evaluación neurofisiológica el patrón clásico corresponde a SNAP de amplitud disminuida (o ausentes), sin compromiso de los parámetros temporales como latencias y velocidad de conducción.
Estas alteraciones se superponen en el mismo nervio con neuroconducción motora normal o levemente alterada y la electromiografía puede ser normal o puede mostrar potenciales de denervación de mínima entidad y/o activación disminuida por pérdida del input sensitivo 1,3,6.
El objetivo de este trabajo es la presentación de un caso evaluado en nuestro servicio, desde el punto de vista clínico y neurofisiológico.
Caso clínico
Paciente de sexo femenino, 53 años, tabaquista intensa, EPOC con diagnóstico de Carcinoma Broncopulmonar de tipo Oat Cell en julio de 2017, en estadío limitado.
Recibió tratamiento oncoespecífico en base a quimioterapia con Cisplatino y Etopósido en Julio, Agosto y Setiembre de 2017 y radioterapia holoencefálica profiláctica que completó en Febrero de 2018 (recibió 2 sesiones de 25 Gy cada una).
En Mayo de 2017, previo al diagnóstico de su enfermedad oncológica comienza con un trastorno de la marcha con arreflexia y apalestesia difusas, interpretado como una Ataxia Sensitiva sin déficit motor significativo.
Luego del inicio de la quimioterapia presentó una peoría rápida de sus manifestaciones neurológicas dada prinicipalmente por dolor de ambos miembros inferiores, sin mejoría luego de culminado dicho tratamiento.
El cuadro fue progresivo, simétrico y comprometió en la evolución miembros superiores, agregando parestesias difusas, llegando a una astasia abasia en 6 meses aproximadamente.
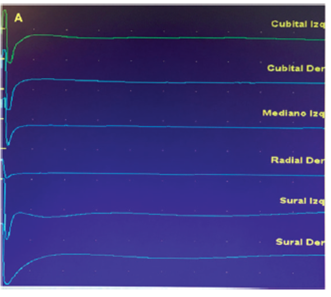
Realizamos el Estudio Eléctrico de los cuatro miembros en Enero de 2018 y constatamos la ausencia de todos los potenciales de acción sensitivos nerviosos en todos los nervios explorados tanto en miembros inferiores como superiores (ambos Nervios Surales, Nervio Radial derecho, ambos Nervios Cubitales y ambos Nervios Medianos), (Figura 1). La neuroconducción motora en miembros superiores (ambos Nervios Medianos y Cubitales) fue normal y en miembros inferiores las alteraciones fueron mínimas (Figura 2).
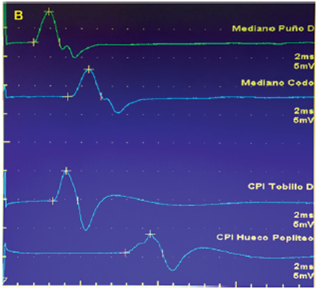
En ambos Nervios Ciáticos Poplíteos Internos el único parámetro mínimamente alterado fue la latencia motora distal a izquierda que fue de 6.3 ms (normal: ≤ 5.8 ms)
En el Nervio Ciático Poplíteo Externo (CPE) izquierdo se registraron amplitud y velocidad de conducción levemente disminuidas, 1.93 mV (normal ≥ 2 mV) y 38 m/s respectivamente (normal ≥ 40 m/s).
La electromiografía mostró alteraciones discretas compatibles con denervación axonal, dadas por actividad espontánea a formas de potenciales de denervación, fibrilaciones y ondas agudas positivas tanto en músculos de miembros superiores como inferiores.
Se completó la evaluación neurofisiológica con la obtención del reflejo T y se comparó con un control sano.
A nivel del reflejo maseterino la respuesta fue normal y a nivel del músculo bíceps no se obtuvo respuesta, lo cual traduce la indemnidad de las fibras Ia aferentes del reflejo maseterino y la alteración de las fibras Ia aferentes de los reflejos osteotendinosos de miembros (en este caso reflejo bicipital), lo cual es característico de las ganglionopatías como fue mencionado anteriormente (Figura 3).
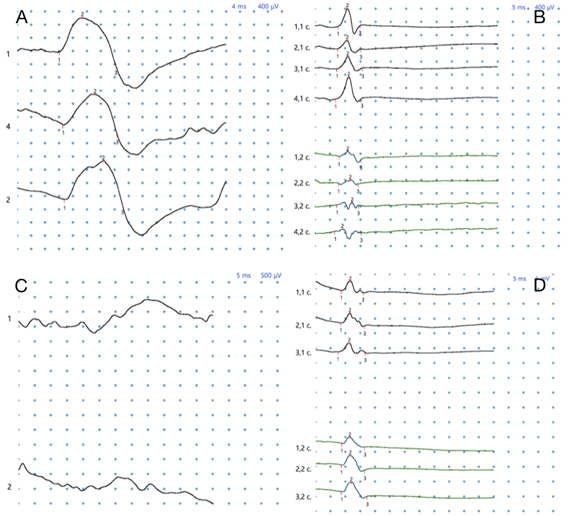
Figura 3: A) Reflejo Bicipital bilateral en control sano. B) Reflejo Maseterino bilateral en control sano. C) Reflejo Bicipital ausente en el paciente. D) Reflejo Maseterino conservado bilateralmente en el paciente.
Se planteó una ganglionopatía paraneoplásica posiblemente agravada por la quimioterapia (Cisplatino) y se indicó tratamiento con bolos de Metilprednisolona 1 gr. intravenoso diario por 5 días. No hubo respuesta a dicho tratamiento por lo que se realizó tratamiento con Inmunoglobulina 500 mg/kg/día por 5 días sin mejoría.
La paciente fue dada de alta sin respuesta a los tratamientos realizados para continuar con control ambulatorio oncológico y neurológico.
Discusión
Como se mencionó, las ganglionopatías se deben a causas adquiridas y hereditarias. Dentro de las primeras se incluyen las paraneoplásicas, autoinmunes, tóxicas, infecciosas e idiopáticas.
Una de las causas más importantes es la paraneoplásica, también denominada neuronopatía sensitiva carcinomatosa y que forma parte de los síndromes paraneoplásicos clásicos.
Se instala en forma subaguda con ataxia sensitiva, asimétrica, de predominio en miembros superiores, asociando dolor y disestesias.
Su planteo debe ir seguido de la búsqueda de neoplasia la cual puede ser diagnosticada en forma concomitante o en la evolución por lo que debe hacerse una búsqueda sistemática de neoplasia que puede repetirse con una frecuencia y duración variable 1,3-5.
La naturaleza de la lesión es inmunomediada y compromete los ganglios de la raíz dorsal debido a la laxitud de la barrera hematonerviosa en este sector, lo que facilita el pasaje de los anticuerpos implicados en la lesión inflamatoria.
La neoplasia más frecuentemente implicada es el cáncer broncopulmonar en su forma oat cell (hasta en el 80%) y los anticuerpos relacionados son el anti-Hu (en la mitad de los casos) del cual existen varios subtipos y que provocan una respuesta inflamatoria mediada por células T CD8+.
Menos frecuentemente los anticuerpos vinculados son el anti-CRMP-5/CV2, anti-YO, anti-anfifisina y en muchos casos no se identifican anticuerpos onconeuronales 3,4.
Otras neoplasias vinculadas son el cáncer de mama, útero, ovario, gastrointestinal, testículo, vejiga, próstata y carcinoma hepatocelular 3.
En algunos casos el daño puede exceder al ganglio sensitivo y extenderse a raíces, columnas posteriores, troncos nerviosos, sistema nervioso autónomo y sistema nervioso central 1,5.
Dentro de las causas autoinmunes la más frecuente es la vinculada a la Enfermedad de Sjögren que origina un cuadro severo de ataxia, arreflexia y disautonomía 1,3.
Menos frecuentemente se relaciona a la Enfermedad Celíaca que se manifiesta por un cuadro sensitivo deficitario multifocal con o sin ataxia.
Casos excepcionales se han relacionado a Hepatitis autoinmune, Lupus Eritematoso Sistémico, Vasculopatía Livedoide y a Enfermedad por anticuerpos antireceptor 3 de factor de crecimiento fibroblástico (anti-FGFR3) 3.
La neuronopatía sensitiva tóxica generalmente es subaguda y se vincula al uso de platinos (cisplatino, carboplatino y oxiplatino) o a Piridoxina.
En el primer caso la toxicidad es dosis dependiente con un umbral de dosis acumulada de 300 mg/m2 y puede asociar polineuropatía periférica. El inicio de las manifestaciones clínicas puede ser diferido hasta 2 meses luego de culminado el tratamiento y la recuperación es lenta y generalmente incompleta 1,3.
La megavitaminosis piridoxínica origina un cuadro de inicio distal con parestesias, inestabilidad en la marcha y posible compromiso autonómico. Aparece con dosis tóxicas de vitamina B6 de 200 mg/día por períodos prolongados y la mejoría es lenta. Ocasionalmente se puede desarrollar un cuadro severo e irreversible debido a necrosis neuronal (1, 3).
Se describen algunos agentes infecciosos asociados ocasionalmente al desarrollo de ganglionopatía como el VIH y menos frecuentemente se han relacionado casos al virus de Ebstein Barr, Varicela Zóster, HTLV-1 y Lepra 3.
Excluídas todas las causas descritas se plantea la etiología idiopática que alcanza en algunas series el 50% y que habitualmente es de evolución lentamente progresiva 3.
Las causas hereditarias son poco frecuentes, pueden ser dominantes o recesivas y se incluyen varias dentro de este grupo 1,3.
La principal manifestación de estas entidades es el déficit sensitivo que puede ser de instalación aguda o crónica. En el primer caso la distribución generalmente es difusa incluyendo el territorio de distribución del V par craneano y en el segundo el inicio es habitualmente distal con expansión centrípeta 1. En ambos casos una vez instalado el cuadro en forma completa el perfil clínico de las manifestaciones sensitivas es no largo dependiente 6.
Habitualmente el déficit se traduce en ataxia sensitiva severa que puede asociar seudoatetosis distal y al examen físico se constata arreflexia difusa. En la evaluación de las fuerzas no se constata déficit, sin embargo ocasionalmente puede evidenciarse una fluctuación en el mantenimiento sostenido de las fuerzas debido al déficit propioceptivo. Estas manifestaciones traducen el compromiso de las neuronas sensitivas de mayor tamaño 1.
En otros casos menos frecuentes la presentación clínica es con déficit termoalgésico, dolor y disestesias, con examen de la sensibilidad propioceptiva y táctil normal. Estas alteraciones traducen el compromiso de las neuronas sensitivas de menor tamaño 1.
El patrón evolutivo es fundamental en la búsqueda etiológica y va desde la desaferentización total en 2 a 3 días hasta cuadros de evolución indolente durante décadas, definiéndose 3 patrones fundamentales, agudo, subagudo y crónico 1.
La forma aguda o neuronopatía sensitiva aguda alcanza su máxima expresión en 72 horas, es de distribución difusa incluyendo el territorio del trigémino y los SNAP están ausentes en el estudio de neuroconducción. La recuperación depende de la eventual compensación del déficit sensitivo por otras vías como la visual y/o vestibular 1.
Las formas subagudas se instalan en semanas a pocos meses y las formas de evolución crónica se instalan en años y generalmente son hereditarias.
Es fundamental el reconocimiento precoz de ésta patología, especialmente para realizar un diagnóstico etiológico sin demora, que permita un tratamiento adecuado con vistas a mejorar el pronóstico 2.
Además de la búsqueda etiológica los estudios paraclínicos tienen el objetivo de distinguir entre ganglionopatía y otras neuropatías sensitivas, para lo cual es fundamental la neurofisiología 3.
El perfil de alteraciones que se observan en el electrodiagnóstico es particular y está definido por anomalías en la neuroconducción sensitiva de perfil no largo-dependiente.
Las mismas en general son severas y difusas, comprometiendo los cuatro miembros aunque pueden predominar en los superiores y es típica la multifocalidad 3,4.
Los SNAP disminuyen de amplitud de forma multifocal (o están ausentes), sin compromiso de los parámetros temporales como latencias y velocidad de conducción. La disminución de la amplitud en los casos subagudos puede ser progresiva con estabilización hacia el décimo mes de evolución. En algunos casos los potenciales sensitivos desaparecen (1,3,6.
Estas alteraciones se superponen en el mismo nervio con neuroconducción motora normal o levemente alterada. La electromiografía puede ser normal, puede constatarse activación disminuida por pérdida del input sensitivo o pueden verse potenciales de denervación de mínima entidad 1,3.
No hay criterios uniformes para el diagnóstico de esta entidad desde el punto de vista de la neurofisiología y los propuestos no son aceptados por todos los autores 7.
Se han descrito criterios de screening neurofisiológico para esta patología como son la presencia de 2 asimetrías de por lo menos 50% en la amplitud de los potenciales sensitivos de un nervio y su homólogo contralateral, lo cual tendría una sensibilidad del 97.1% y una especificidad del 94.1%.
Si las asimetrías fueran en 3 nervios la especificidad aumenta al 100% y si la asimetría alcanza al 100% la sensibilidad es del 100% 2.
Se plantean otros parámetros y técnicas útiles en la evaluación de las ganglionopatías como la Razón de amplitud sensitivo/motora del Nervio Cubital, la comparación del registro del reflejo H con evocación proximal y distal 2 y el registro del reflejo maseterino o reflejo T 8.
En el primer caso la razón de amplitud sensitivo/motora Cubital de 0.71 o menos tiene una sensibilidad del 94.4% y una especificidad del 90.9% para ganglionopatía 6.
Como mencionamos desde el punto de vista clínico, el cuadro se asocia a una arreflexia difusa, sin embargo el reflejo maseterino está conservado.
Esto se debe a que el cuerpo neuronal de la neurona sensitiva propioceptiva se encuentra a nivel del núcleo sensitivo protuberancial del trigémino (neurona aferente del reflejo) y no en el ganglio de Gasser. Esto se pone en evidencia en la neurofisiología con el registro del reflejo T, el cual se registra mediante el uso del martillo de reflejos del electromiografo que actúa como estímulo y registrando el potencial de acción muscular compuesto en el músculo masetero.
Si utilizamos la misma técnica para el resto de los reflejos osteotendinosos no se obtendrá registro de potencial de acción muscular compuesto 8,9.
Debe realizarse una búsqueda sistemática de neoplasia mediante técnicas de imagen y dosificación de anticuerpos onconeuronales, principalmente el anti-Hu e incluso puede considerarse la realización de tomografía por emisión de positrones con fluorodesoxiglucosa que puede repetirse cada 6 meses por 2 años 4.
En el caso de la ganglionopatía de origen paraneoplásico el tratamiento debe ser oncoespecífico en primer lugar asociando tratamiento inmunomodulador con corticoides o inmunoglobulinas intravenosas. Es fundamental tener presente la corta ventana terapéutica ya que la lesión inflamatoria rápidamente lleva a la muerte neuronal y a la lesión irreversible, como ocurrió en nuestra paciente 3.
En los cuadros de naturaleza autoinmune el tratamiento fundamental es inmunosupresor y se plantea el uso de corticoides, micofenolato, azatioprina, inmunoglobulinas y plasmaféresis entre otros 3.
Es fundamental el reconocimiento precoz de ésta patología, especialmente para realizar un diagnóstico etiológico sin demora, que permita un tratamiento adecuado con vistas a mejorar el pronóstico 2.

