











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
La esclerosis sistémica(ES) es una enfermedad crónica y heterogénea caracterizada por fibrosis generalizada de la piel y órganos internos, vasculopatía de pequeños vasos, asociada a la producción de autoanticuerpos. Afecta principalmente mujeres entre la cuarta y quinta década de la vida aumentando su incidencia con la edad 1) (2.
Como toda afección sistémica, su forma de presentación es muy variada, dependiendo del tejido mayormente afectado. Dentro de las manifestaciones clínicas fácilmente observables se destaca la induración progresiva de la piel con compromiso variable de cara, pudiendo aparecer telangiectasias faciales, hiperpigmentación y calcificaciones cutáneas. El fenómeno de Raynaud (FR) con diverso grado de severidad, pero frecuentemente con dolor y úlceras digitales, suele acompañar a esta enfermedad. También aparecen artralgias de diversa entidad y compromiso de órganos internos. Dentro de éstos últimos, el compromiso respiratorio marca la gravedad y el pronóstico, junto con la Hipertensión Pulmonar. El tubo digestivo suele comprometerse, provocando frecuentemente reflujo gastroesofágico severo 3) (4.
Según la extensión del compromiso cutáneo se puede clasificar a la enfermedad en:
5) 6). Asimismo, existe un subtipo de pacientes con ES temprana, también denominada “pre-esclerodermia”, que se caracteriza por presentar alteraciones capilaroscópicas, anticuerpos antinucleares positivos y FR. Estos pacientes son considerados de alto riesgo para desarrollar en la evolución una ES definida 7.
El tratamiento de la enfermedad suele estar poco sistematizado, ensayándose diversas terapéuticas con niveles de evidencia variables. Las guías internacionales constituyen una fuerte orientación para la práctica clínica pero deben ajustarse a las realidades locales, por lo que conocer la situación en determinado lugar puede permitir implementar mejoras en el manejo de esta difícil entidad.
En nuestro medio se han presentado series que muestran experiencias locales sobre esta interesante y desafiante enfermedad 8. Sin embargo, disponemos de escasos datos nacionales y regionales de prevalencia de la enfermedad, pero no de la frecuencia de las formas de presentación. A su vez, tampoco conocemos el manejo terapéutico que se realiza en nuestro entorno de esta enfermedad, que como se menciona más arriba, es un tema controvertido y no siempre sistematizado.
El presente trabajo tiene por objetivo aportar información que permita conocer la forma de presentación de esta enfermedad y el manejo terapéutico de la misma, de acuerdo a las manifestaciones clínicas involucradas.
Material y métodos
Se realizó un corte transversal de pacientes con diagnóstico de ES, clasificados según las recomendaciones ACR/EULAR 2013. Se incluyeron todos los pacientes asistidos y en seguimiento por nuestro Grupo de Trabajo en Enfermedades Autoinmunes Sistémicas desde 2013 a la fecha. Este grupo incluye hasta el momento tres Unidades pertenecientes a dos Instituciones de Asistencia Médica Colectiva privadas y una perteneciente a un Hospital público nacional donde funcionan servicios docentes de la Facultad de Medicina de la Unidad de la República. En total, estos tres centros asisten 600 mil usuarios. Todas funcionan en Montevideo, Uruguay y son de referencia para otras especialidades y zonas geográficas del país.
Se revisaron las Historias Clínicas de los pacientes mediante un formulario precodificado de recolección de datos. Los pacientes con diagnóstico de ES fueron obtenidos de nuestra base de datos.
De cada caso se analizó las siguientes variables: edad, sexo, perfil clínico, manifestación clínica predominante, comorbilidades, autoanticuerpos, capilaroscopía, tratamiento inmunosupresor recibido en los últimos 6 meses, uso de glucocorticoides, motivo de indicación y efectos adversos.
Para identificar los perfiles clínicos se siguieron los criterios de LeRoy y Medsger:
-ES difusa, la esclerosis cutánea se extiende proximal a los codos, rodillas o incluye tronco.
-ES limitada, la esclerosis cutánea está confinada a la parte distal a los codos, rodillas y/o la cara.
-ES sin esclerodermia definida por la presencia de FR, compromiso visceral compatible con ES, ausencia de endurecimiento cutáneo y positividad para los Anticuerpos Antinucleares (ANA).
A los efectos de los compromisos orgánicos se consideró: rigidez/endurecimiento cutáneo objetivado por un médico del Grupo, medido por Score de Rodnan modificado (mRSS), hipertensión pulmonar objetivada por Ecocardiograma (presión sistólica de la arteria pulmonar mayor a 35 mmHg) Enfermedad Pulmonar Intersticial (EPI) objetivada por Tomografía de Tórax de alta resolucióny estudio funcional respiratorio con difusión de monóxido de carbono (CO) (capacidad vital forzada menor de 70% o difusión de CO menor de 80%), reflujo gastroesofágico objetivado por estudios radiológicos contrastados.
El protocolo de investigación fue aprobado por las Instituciones participantes y se cumplieron los requisitos de la Declaración de Helsinki. Los datos categóricos son expresados como proporciones y los cuantitativos con su frecuencia absoluta y como mediana y rango. Los datos se procesaron con el programa estadístico SPSS 21.0.
Resultados
Se analizaron 49 casos de ES. La mediana de edad fue 63 años (25-85), 44 (90%) de sexo femenino.
El perfil clínico fue: 5 (10%) ES sin esclerodermia, 19 (39%) ES difusa, 25 (51%) ES limitada.
La manifestación clínica predominante fue: endurecimiento cutáneo en 42 (85,7%) casos, FR 41 (83,7%), articular 27 (55,1%), tubo digestivo 24 (49%), EPI 16 (32,7%), hipertensión pulmonar estimada por Ecocardiograma 7 (14,3%), renal 3 (6,1%) y otros 2 (4,1%).
Se constataron factores de riesgo cardiovascular diferentes a tabaquismo en 12 casos (24,5%) y tabaquismo en 7 casos (14,3%). Las manifestaciones clínicas según el perfil clínico se presentan en la Tabla 1.
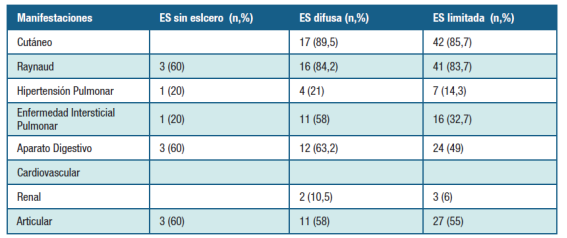
El perfil de autoanticuerpos mostró que 39 (79,6%) casos tenían Anticuerpos Antinucleares (ANA) positivos, en 11 (22,4%) anticuerpos anticentrómero, 3 (6,1%) antitopoisomerasa I o Scl-70, anti-RNP en 4 (8,2%) casos, otros autoanticuerpos (anti-Ro, anti-La, factor reumatoideo) en 13 (26,5%) casos. La relación entre los autoanticuerpos y el perfil clínico se presentan en la Tabla 2.
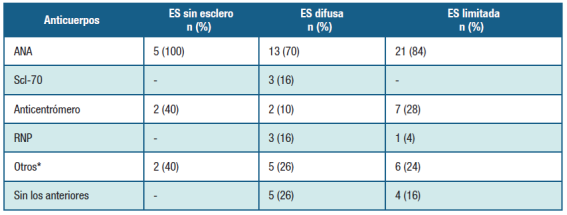
* Puede ser concomitantes. ANA: anticuerpos antinucleraes; Scl-70: anticuerpos anti Scl-70; RNP: anticuerpos antiribonucleoproteina. Nota: no se discriminaba al momento de este estudio el tipo de RNP.
La capilaroscopía estuvo informada como patrón predominante con megacapilares en 15 (30,6%) casos, áreas avasculares en 16 (32,6%), mixto en 10 (20,4%) y sin realizar en 8 (16%) casos.
Recibieron tratamiento inmunosupresor 17 (34,6%) de pacientes, siendo el motivo de inmunosupresión en 8 (16,3%) casos el compromiso respiratorio, en 5 (10,2%) casos el compromiso cutáneo y en 4 (8,2%) casos el compromiso articular. En todos los casos, el tratamiento se indicó como tratamiento de primera instancia. Este tratamiento inmunosupresor se empleó más frecuentemente en la ES difusa (10 casos, 20,4%).
Los medicamentos empleados para la EPI fueron Ciclofosfamida en 8 casos (tratamiento de inducción), Micofenolato de Mofetilo (5/8) y Azatioprina (3/8) para tratamiento de consolidación y mantenimiento. Recibieron prednisona 23/49 pacientes. La causa más frecuente de uso de Prednisona fue el compromiso articular, en 15/49 casos. No se constataron efectos adversos que motivaran suspensión del tratamiento ni determinaran ingresos hospitalarios.
Discusión
Nuestro trabajo permite conocer algunos datos locales en relación a la forma de presentación y manejo terapéutico de la ES. Se trata de una cohorte de pacientes con una mediana de edad mayor al promedio de edad de debut de esta enfermedad. Esto seguramente es atribuible a que se trata de un estudio transversal de pacientes que ya vienen en seguimiento. Sin embargo, el resto de las variables demográficas y la forma de presentación clínica es comparable a la observada en los registros de numerosos pacientes con ES, como el Registro Español de ES (RELES)9.
En efecto, en nuestra serie la relación mujer: hombre fue 9:1, predominó la forma limitada de la enfermedad, seguida de la forma difusa y de la presentación sin esclerodermia.
Cabe agregar que en todas las series la forma limitada suele ser diagnosticada a edades más avanzadas, por lo que el predominio de esta forma de presentación puede influir en la edad avanzada que encontramos en este estudio 9,10.
Las manifestaciones clínicas observadas en nuestra serie también muestran frecuencias similares a las halladas en otras. El endurecimiento cutáneo y el FR se observaron en más de 80% de los casos, seguido por el compromiso articular, manifestaciones digestivas y EPI. El FR suele estar presente en la mayoría de los pacientes con ES, asociándose a una evolución adversa, como la presencia de fibrosis de dedos, perdida de pulpejos, ulceras y amputación digital. En algunos casos, el FR antecede a las manifestaciones típicas de ES, pero su asociación con una capilaroscopía sugestiva y un perfil de autoanticuepos compatible, aumentan claramente la probabilidad de desarrollar la enfermedad 11. Tal como se ha observado en otras series, observamos que la EPI está claramente más representada en la forma difusa 9) (10.
La afección pulmonar es prevalente en la ES, siendo las complicaciones a este nivel la principal causa de mortalidad. Hay dos formas fundamentales de afección pulmonar, la EPI y la hipertensión arterial pulmonar (HAP) 12. La EPI es más frecuente, detectada hasta en 40% de los pacientes con ES. Las alteraciones más tempranas deben evidenciarse por una disminución en la difusión del monóxido de carbono (DLCO) en las pruebas funcionales. Este estudio es el mejor indicador para estimar el daño pulmonar, puesto que es más sensible que los estudios imagenológicos 12.
El compromiso renal fue muy limitado en nuestra serie. Actualmente, el compromiso renal es raro. La introducción de los inhibidores de la enzima de conversión de angiotensina (IECA) ha descendido su incidencia pero sigue constituyendo una complicación potencialmente fatal, siendo la crisis renal esclerodérmica la presentación característica. Los principales factores de riesgo para el desarrollo de esta complicación son el uso previo de corticoides, el compromiso cutáneo difuso y rápidamente progresivo y la presencia de anticuerpos anti RNA polimerasa III 13.
El análisis de los autoanticuerpos reveló la presencia de ANA en 80% de los casos. Dentro de los anticuerpos dirigidos contra antígenos extraíbles nucleares encontramos anticuerpos anticentrómero, predominando en la forma limitada. Los anticuerpos antitopoisomerasa I fueron observados con baja frecuencia. Si bien es conocido que estos últimos autoanticuerpos, se encuentran con baja frecuencia en la ES, nuestra observaciónes llamativamente baja.
Otras series han reportado presencia de anticuerpos antinucleares (ANA) en un 90-100% de los casos. Los anticuerpos antitopoisomerasa I (Scl-70) suelen estar presentes en menos de la mitad de los casos, habitualmente en pacientes con la forma difusa de la enfermedad y asociados al desarrollo de compromiso intersticial pulmonar. Su presencia indica un peor pronóstico. Los anticuerpos anti-centrómero se ven en un tercio de los casos y se asocian a la forma limitada de la enfermedad así como a la HAP 13,14. Las discordancias en las frecuencias observadas con otras series sugieren que existen inconvenientes en la técnica de detección de los autoanticuerpos en nuestro medio, que deberán ser corregidos. En efecto, como corolario de este estudio se desprenden debilidades internas que deben ser corregidas para mejorar la calidad asistencial de estas patologías por nuestro grupo.
Los hallazgos capilaroscópicos también merecen ser apreciados. Actualmente la capilaroscopía constituye una herramienta útil en el diagnóstico y seguimiento de éstos pacientes. Se han descrito tres patrones esclerodermiformes: avascular, megacapilares y mixto, planteándose la correlación entre algunos de estos patrones y la evolución clínica de la enfermedad 11. Es de destacar que en nuestra serie, en la forma difusa se observó un predominio del patrón avascular, también conocido como patrón activo, al tiempo que en la forma limitada y en la ES sin esclerodermia predominó el hallazgo de megacapilares, también conocido como patrón de lenta progresión.
El uso de medicamentos inmunosupresores predominó en pacientes con compromiso respiratorio y en la forma difusa, lo que es compatible con la mayor presencia de EPI en esta forma de presentación. En estos casos se empleó Ciclofosfamida, Micofenolato de Mofetilo y Azatioprina. El uso de Ciclofosfamida está claramente establecido en la literatura para el manejo de la EPI asociada a la ES, mientras que los datos para Micofenolato y Azatioprina son menos concluyentes 15,16. Nuestra casuística es pequeña en relación a la presencia de EPI, pero aun así se observa una dispersión en la terapéutica, pasible de ser protocolizada.
Está bien establecido que los pacientes con EPI requieren tratamiento inmunosupresor. En la literatura suele emplearse Ciclofosfamida, mientras que Azatioprina y Micofenolato de Mofetilo pueden ser considerados una alternativa razonable 15-17. Actualmente el uso de Rituximab constituye una alternativa en pacientes refractarios al tratamiento convencional, habiéndose reportado resultados alentadores con detención de la progresión de la enfermedad pulmonar e incluso mejoría de los parámetros del estudio funcional respiratorio 18,19. En nuestro medio el acceso a este medicamento es escaso por lo que la experiencia es casi nula.
El tratamiento del compromiso cutáneo es complejo y está menos sistematizado. Se ha utilizado Metrotexate, Ciclofosfamida, Micofenolato e inclusive Rituximab con resultados variables. El uso de antifibróticos se encuentra en fase de experimentación 20.
Una mención especial cabe para el manejo del FR severo, que ha requerido incorporar al tratamiento clásico (calcio antágonistas, inhibidores de la fofodiesterasa), los antagonistas de la endotelina 1 (Bosentan) con el objetivo de reducir el número, duración y gravedad de las crisis así como la prevención de ulceras digitales 21) (22.
A su vez, el uso de glucocorticoides predominó en el compromiso articular. Es bien conocido que el uso de este grupo terapéutico debe ser juicioso por los numerosos efectos adversos que puede determinar. En particular, en el caso de la ES, se agrega el riesgo de agravar el compromiso renal y precipitar una crisis renal esclerodérmica. Por esta razón, la dosis de prednisona o equivalente no debe ser superior a los 10 a 15 mg/día, debiéndose recurrir precozmente a ahorradores de corticoides, como Metotrexate 20.
Nuestro estudio tiene limitaciones diversas, derivadas del diseño y del número limitado de casos. Sin embargo, conocer datos locales y, en particular el perfil clínico y el manejo terapéutico que hacemos de nuestros pacientes permite hacer un control de calidad de nuestra propia asistencia. La fortaleza más importante deriva de que se trata de nuestros pacientes. Es una serie basada en pacientes reales y no de grandes registros que aportan datos valiosos pero de pacientes desconocidos para el equipo investigador. Es necesario sistematizar y ampliar el registro y la realización de bases de datos para poder tener datos epidemiológicos locales confiables y útiles en las decisiones clínicas. A su vez, nuestro trabajo pone en evidencia la necesidad de protocolizar los tratamientos de esta enfermedad, compleja y de baja frecuencia, para que sea más sistemático, basado en la evidencia y, en consecuencia, menos librado al criterio de cada profesional actuante.
Los autores no tienen conflictos de interés para declarar en relación al presente estudio.

