











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
El término ataxiasignifica desde el punto de vista clínico, inestabilidad e incoordinación. Sin embargo, también es utilizado para definir a un amplio grupo de enfermedades del sistema nervioso central, en las cuales ataxia global y progresiva es el síntoma predominante(1, 2, 3).
Están excluidas de este grupo las etiologías focales o estructurales que pueden generar ataxia (tumorales, infecciosas, vasculares y desmielinizantes). Estas entidades se presentan con cuadros muy asimétricos o unilaterales, a diferencia de las ataxias globales (de aquí en adelante “ataxias”) donde los cuadros suelen ser más homogéneos, si bien las asimetrías son también posibles.
Comprenden un grupo heterogeneo de enfermedades de origen tanto genético como adquirido 1),(3. La frecuencia de las mismas es desconocida, pero se estima que existen entre 50 y 100 tipos de ataxias 1.
Con un criterio etiológico, las ataxias se clasifican en tres grupos principales: ataxias adquiridas, ataxias hereditarias y ataxias degenerativas no genéticas 1. Realizar un correcto diagnóstico etiológico es un desafío debido a la superposición fenotípica y a la variabilidad en la forma de presentación de las diferentes nosologías 1,2,4.
El cuadro clínico y los antecedentes familiares nos pueden orientar en la búsqueda etiológica. Por ejemplo, en pacientes jóvenes cuya enfermedad comienza antes de los 25 años y afecta solo a una generación familiar, el diagnostico mas probable es una ataxia autosómica recesiva y dentro de ellas, por frecuencia, una ataxia de Friederich5,6. Si uno de sus padres tiene una afectación similar, una ataxia espinocerebelosa (SCA, por su sigla en inglés) es el diagnóstico más probable (1,3,4.
Los métodos de imágenes como la resonancia magnética (RM), colaboran en la caracterización de estos desordenes, no obstante, el diagnóstico definitivo muchas veces se basa en una combinación de test bioquímicos y genéticos 2,4. En algunas ocasiones, los hallazgos extra neurológicos facilitan dicha caracterización.
Sin embargo, lo más habitual y que a su vez plantea un mayor desafío diagnóstico al neurólogo, es cuando la ataxia se manifiesta en el adulto, generalmente mayor de 35 o 40 años (aunque no hay un criterio de edad definido), sin una historia familiar de ataxia. Es a este grupo de pacientes que se les denomina, con un criterio nosológico, como portadores de una ataxia esporádica de comienzo en el adulto (AECA). En estos pacientes casi todo el espectro de causas genéticas y no genéticas deben ser consideradas en la búsqueda etiológica1.
La historia clínica, el examen neurológico, extraneurológico y las características imagenológicas, son cruciales para guiar las investigaciones y acotar la lista de los posibles diagnósticos.
Estos pacientes pueden tener una ataxia adquirida, una ataxia degenerativa esporádica y hasta un 20% de ellos, pueden tener una mutación genética responsable del cuadro clínico, a pesar de tener una historia familiar negativa. Por este motivo, es que se recomiendan las pruebas moleculares para ataxia de Friedreich y SCA cuando no hay una clara evidencia etiológica 4.
Cuando los estudios iniciales no permiten llegar al diagnóstico de una etiología específica o condición tratable, la posibilidad de una condición degenerativa primaria o hereditaria adquiere más importancia 3.
En las últimas décadas se han producido grandes avances en el campo de la genética y de las alteraciones moleculares vinculadas a las ataxias. Además, la definición de criterios clínicos e imagenológicos en diversas entidades nosológicas, ha facilitado un correcto diagnóstico en numerosas formas de ataxias esporádicas. A pesar de todo esto, poder llegar a un diagnóstico etiológico en las AECA, continúa siendo un gran desafío para el neurólogo 3.
En nuestro país no es fácil el acceso a los análisis genéticos para el estudio etiológico de las AECA, por lo tanto, es muy difícil confirmar el diagnóstico de las distintas ataxias hereditarias, y a su vez, discriminar entre estas y las ataxias degenerativas adquiridas, salvo con la atrofia multisistémica (AMS) que posee criterios diagnósticos específicos7.
Este es uno de los motivos por lo que enfocamos nuestro estudio en las AECA de causa adquirida. También justificamos la realización de este trabajo, dada la ausencia en nuestro país de estudios sobre esta entidad, desconociéndose la frecuencia de cada nosología responsable de la misma. La etiología adquirida posee además un especial interés, dado la posibilidad de mejoría o estabilización del cuadro clínico con el tratamiento especifico. Además, conocer las características clínicas y/o imagenológicas de las diferentes entidades, nos puede orientar en la solicitud de estudios paraclínicos en busca de una etiología específica.
El objetivo primario del presente trabajo es describir las características clínicas e imagenológicas de los pacientes con AECA; y el objetivo secundario es describir la frecuencia y las características clínicas e imagenológicas de las diferentes etiologías de las AECA adquiridas.
Material y métodos
Se trata de un estudioprospectivo y descriptivo, en el que se incluyeron todos los pacientes valorados por AECA de 35 años o más, que consultaron en la policlínica de neurología general o fueron ingresados en sala de neurología del Hospital de Clínicas, en el período comprendido entre abril de 2010 y marzo de 2014.
Utilizamos este criterio etario en forma arbitraria ya que los diferentes autores no coinciden en una edad específica de comienzo de los síntomas, algunos utilizan como punto de corte 35 años y otros 40 años.
Los criterios de inclusión son: inicio de la ataxia a una edad mayor o igual a los 35 años, ausencia de antecedentes familiares de ataxia, pacientes en seguimiento en policlínica de neurología general o ingresados en sala de neurología del Hospital de Clínicas.
Los criterios de exclusión fueron: comienzo de los síntomas antes de los 35 años, presencia de antecedentes familiares de ataxia, causa estructural (vascular, tumoral o desmielinizante) causante del cuadro clínico evidenciada por imagenología.
Ataxia adquirida: pacientes en los que se encontró una noxa adquirida que explicara el cuadro clínico
Ataxia no adquirida: pacientes en los que no se encontró una noxa adquirida que explicara el cuadro clínico, los cuales pueden corresponder a una noxa degenerativa, hereditaria o adquirida pero que no cumpliera con los criterios diagnóstico al momento de finalizar el estudio
En los pacientes que consultaron por primera vez en este período de tiempo, se realizó evaluación clínica y se solicitaron los estudios paraclínicos necesarios para la búsqueda etiológica.
En los pacientes que concurrieron a control en este período de tiempo, se realizó una nueva evaluación clínica, se revisó su historia clínica y se solicitaron estudios paraclínicos no solicitados previamente o que era necesario repetir.
El algoritmo de solicitud de los estudios paraclínicos fue el siguiente:
Imagenología de cráneo: Resonancia Magnética(RM).
En caso de no llegar al diagnóstico etiológico, se solicitaron los siguientes exámenes:TSH, HIV, VDRL, anticuerpos antitiroideos, tomografía (TC) de tórax, abdomen y pelvis, dosificación de vitamina B12, dosificación de ácido fólico, ANA, VES y anticuerpos para enfermedad celíaca.
En caso de negatividad de los mismos u orientación clínica, se solicitó alguno de los siguientes, según cada caso individual: mamografía, PAP, anticuerpos onconeuronales, marcadores oncológicos (PSA, CA 19-9, CEA 125, CEA), RM de médula, EEG, EE, SPECT, PET, y estudio de LCR.
Se recogieron los datos en un protocolo específicamente confeccionado para este trabajo. Se solicito consentimiento informado para el ingreso al estudio.
Resultados
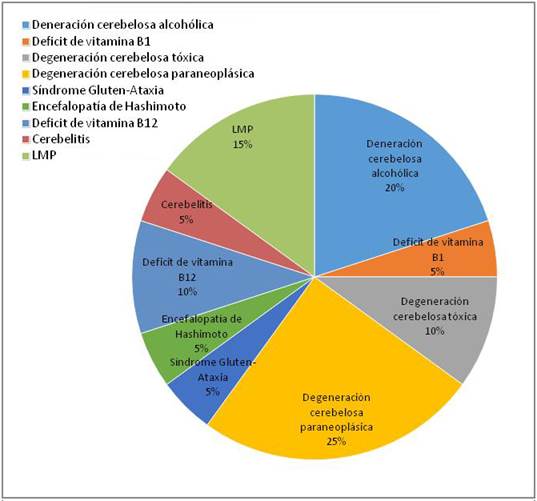
Se incluyeron 36 pacientes con AECA, 20 con etiología adquirida y 16 no adquirida.En éste último grupo, se encontraron 3 pacientes con atrofia multisistémica tipo C (AMS-c), entidad que tiene criterios diagnóstico específicos (7. En la Gráfica 1 se presentan las etiologías adquiridas.
El promedio de edad de comienzo para las AECA fue de 55 años (rango 35 a 92 años), 59 años para los pacientes con etiología adquirida y51 años para los pacientes de etiología no adquirida.
El 61% (22 pacientes) fueron de sexo femenino y 39% (14 pacientes) de sexo masculino considerando todos los pacientes con AECA. Para las causas adquiridas 13 pacientes fueron mujeres y 7 hombres, y para las causas no adquiridas fueron 9 mujeres y 7 hombres. En la Tabla 1 se muestra la presentación clínica y en la Tabla 2 las mismas agrupadas en síndromes.
| Etiología Adquirida | Etiología No Adquirida | Total | |
| Ataxia de la estática | 17 | 15 | 32 (88,9%) |
| Ataxia de la marcha | 18 | 14 | 32 (88,9%) |
| Ataxia de miembros | 12 | 15 | 27 (75%) |
| Disartria | 9 | 10 | 19 (52,8%) |
| Hipotonía | 6 | 8 | 14 (38,9%) |
| Vestibular | 6 | 5 | 11 (30,6%) |
| Temblor | 6 | 3 | 9 (25%) |
| Oculomotricidad | 1 | 3 | 4 (11,1%) |
| Ataxia sensitiva | 2 | 0 | 2 (5,6%) |
| Manifestaciones asociadas | 8 | 12 | 20 (55,6%) |
| Progresión rápida | 4 | 0 | 4 (11,1%) |
| Progresión lenta | 16 | 16 | 32 (88,9%) |
Tabla 1: Manifestaciones clínicas
| Etiología Adquirida | Etiología No Adquirida | Total | |
| Síndrome cerebeloso hemisférico - vermiano | 13 | 15 | 28 (77,8%) |
| Síndrome cerebeloso vermiano puro | 6 | 0 | 6 (16,7%) |
| Síndrome cerebeloso hemisférico puro | 2 | 0 | 2 (5,6%) |
Tabla 2: Manifestaciones clínicas por síndrome
Presentaron síntomas acompañantes 20 pacientes (Tabla 1), los cuales fueron los siguientes: en 10 pacientes manifestaciones piramidales (2 de etiología adquirida y 8 no adquirida), 3 asociaron parkinsonismo (todos con etiologías no adquiridas), 1 paciente asoció mioclonias (fue un síndrome gluten-ataxia), 2 pacientes presentaron disautonomía (ambos con causas no adquiridas) y 10 pacientes asociaron manifestaciones del sistema nervioso periférico (6 de etiología adquirida y 4 de etiología no adquirida).
Respecto al curso evolutivo, el mismo fue rápido en 4 pacientes, todos ellos con causas adquiridas (déficit de vitamina B1, degeneración cerebelosa paraneoplásica definida, cerebelitis viral aguda y LMP). En los otros 32 pacientes la evolución fue lenta (16 con etiología adquirida y 16 no adquirida).
La imagenología fue patológica en 21 pacientes, 9 de los cuales tuvieron una causa adquirida y 12 no adquirida (Tabla 3). En los otros 15 pacientes la imagenología fue normal, teniendo 11 causas adquiridas de AECA y 4 no adquirida. En 18 pacientes se evidenció atrofia cerebelosa difusa, en 6 de los cuales la etiología fue adquirida (1 degeneración cerebelosa alcohólica, 1 degeneración cerebelosa tóxica, 1 degeneración cerebelosa paraneoplásica definida, 1 encefalopatía de Hashimoto y 2 LMP). La atrofia cerebelosa fue a predominio vermiano en 2 pacientes, ambos con causas adquiridas. En 5 pacientes se encontró atrofia de tronco encefálico, todos con etiologías no adquiridas. En 3 pacientes se vio compromiso de la sustancia blanca cerebelosa, todos por LMP.
| Etiología Adquirida | Etiología No Adquirida | Total | |
| Imagen patológica | 9 | 12 | 21 (58,3%) |
| Imagen normal | 11 | 4 | 15 (41,7%) |
Tabla 3: Características imagenológicas.
De los 20 pacientes con AECA de etiología adquirida, 15 recibieron tratamiento etiológico, 8 mejoraron, 5 se mantuvieron estables y en 2 no se obtuvieron datos de respuesta al tratamiento. No recibieron tratamiento etiológico 5 pacientes. De los 8 pacientes que mejoraron en respuesta al tratamiento, 2 tenían déficit de vitamina B12, 1 déficit de vitamina B1, 1 encefalopatía de Hashimoto, 1 síndrome gluten-ataxia, 1 degeneración cerebelosa paraneoplásica definida, 1 cerebelitis aguda y 1 LMP. Los 5 pacientes que se mantuvieron estables, fueron 3 por degeneración cerebelosa alcohólica, 1 por degeneración cerebelosa tóxica y 1 por LMP. Los 5 pacientes que no recibieron tratamiento etiológico empeoraron en la evolución.
Discusión y comentarios
Más de la mitad de los pacientes (56%) con AECA presentaron una etiología adquirida de su ataxia, lo cual implica que puedan tener un tratamiento específico, con una mejoría o detención en el curso evolutivo, dependiendo de la nosología. Las dos etiologías más frecuentes fueron la degeneración cerebelosa alcohólicay la degeneración cerebelosa paraneoplásica, representando casi el 50% de las causas adquiridas8-10.
Como se definió en materiales y métodos, en los 16 pacientes (44%) en los cuales no se encontró una etiología adquirida,esta pueden ser degenerativa o hereditaria, sin poder descartar las etiologías adquiridas no diagnosticadas al momento del estudio.
Al no contar con estudios genéticos en el presente estudio, no se puede lograr discriminar que porcentaje se debió e esta etiología, sabiendo que en algunas series esta situación llega al 20%.
Se encontró una mayor edad promedio de comienzo de los síntomas en las etiologías adquiridas versus las no adquiridas (59 y 51 años respectivamente). Este hecho podría explicarse por la participación de las nosologías genéticas no diagnosticadas en este último grupo, que usualmente comienzan a edades más tempranas.
Como cabía esperar, la ataxia fue el síntoma predominante en los dos grupos, ya sea de la estática, la marcha o de los miembros, siendo menos frecuentes los demás síntomas analizados. La ataxia de miembros se encontró en casi la totalidad de los pacientes con etiologías no adquiridas, en cambio, en las adquiridas se evidenció en una menor proporción. Esta situación puede explicarse por el mayor impacto que tienen en el sector vermiano (y no tanto en los hemisferios cerebelosos) algunas noxas adquiridas tóxicas como la degeneración cerebelosa alcohólica9,10.
Ataxia sensitiva (cordonal posterior) se presentó en solo 2 pacientes y ambos correspondieron a una ataxia adquirida y dentro de ella a un déficit de vitamina B12, por lo que su presencia debería alertarnos sobre la presencia de una etiología potencialmente tratable, específicamente a carencias vitamínicas como así también a neurosífilis (aunque no se haya encontrado ninguna en nuestro trabajo)11.
La asimetría en el síndrome cerebeloso hemisférico solo se observó en 2 pacientes de etiología adquirida (LMP), por lo que su presencia debería hacernos pensar en dicha patología, así como en causas vasculares, tumorales o desmielinizantes, entidades no incluidas en nuestro trabajo12,13.
Un resultado interesante es que cuando se encontró un síndrome vermiano puro, sin acompañarse de un síndrome hemisférico (lo que sucedió en 6 pacientes), todos se debieron a etiologías adquiridas. Por lo tanto, en nuestro estudio, la presencia de un síndrome vermiano puro fue un hallazgoque se observó solo en las causas adquiridas de ataxia.
De los 16 pacientes con etiología no adquiridas, 15 se presentaron con un síndrome cerebeloso completo, hemisférico y vermiano. En las causas adquiridas este hecho se dio en 13 de 20 casos.
Los elementos acompañantes son un elemento importante a la hora de evaluar este tipo de pacientes, dado los aportes que nos pueden brindar en la orientación etiológica. Se presentaron manifestaciones acompañantes en 20 de los 36 pacientes, siendo las más frecuentes los signos piramidales y las manifestaciones de compromiso del sistema nervioso periférico. En este sentido, un hallazgo interesante fue que solo 8 de los 20 pacientes con etiología adquirida los presentaron, a diferencia de las no adquiridas, donde se encontraron en 12 de los 16 pacientes. Estos resultados nos deberían hacer pensar en la posibilidad de una noxa potencialmente tratable cuando nos enfrentemos a un sindrome cerebeloso puro.
Las manifestaciones piramidales, parkinsonianas y disautonómicas predominaron claramente en las etiologías no adquiridas, en cambio, el compromiso del sistema nervioso periférico fue incluso más frecuente en las etiologías adquiridas, por lo que su presencia nos debería hacer pensar en esta nosología (ej: degeneración cerebelosa + polineuropatía por alcohol).
Se debe recordar que las causas degenerativas no hereditarias como la atrofia multisistémica (AMS), así como causas hereditarias como las SCA pueden presentar afectación de múltiples sistemas neurológicos: piramidal, extrapiramidal, autonómico, periférico, visual, etc.
Las mioclonias encontradas en un paciente portador de síndrome gluten-ataxia, son un elemento clínico característico de esta nosología, aunque no específico (se pueden observar también en la Encefalopatía de Hashimoto o en variantes cerebelosas de la Enfermedad de Creutzfeld Jakob)14-19.
Uno de los elementos clínicos más relevantes a la hora de evaluar la probabilidad de una etiología adquirida es el curso evolutivo. En nuestro trabajo quedó claramente demostrado dicha importancia al encontrar que en todos los pacientes en los cuales el curso evolutivo fue rápidamente progresivo, la etiología fue adquirida.
En los últimos años, la imagenología ha adquirido un rol fundamental a la hora de evaluar este tipo de pacientes, posicionándose según la mayoría de los autores, en el primer estudio paraclínico a solicitar ante la presencia de una AECA. Permite además demostrar rápidamente causas estructurales de la ataxia (vasculares, tumorales, desmielinizantes, etc.). Aproximadamente la mitad de los pacientes con etiología adquirida presentó una imagenología de cráneo normal, situación que solo ocurrió en la cuarta parte de los pacientes en los que no se pudo demostrar etiología adquirida.
Las características imagenológicas que se asociaron a una etiología adquirida fueron la atrofia a predominio vermiano y el compromiso de la sustancia blanca cerebelosa, características que no se presentaron en ningún paciente con etiología no adquirida. Una característica a destacar es que los 3 pacientes que presentaron afectación de la sustancia blanca cerebelosa correspondieron a LMP12,13.
La atrofia de tronco encefálico se asoció claramente a etiologías no adquiridas.
Se realizó tratamiento etiológico en el 75% de los pacientes con ataxia adquirida, observando mejoría clínica en más de la mitad y una estabilidad en una tercera parte de los mismos, no encontrándose empeoramiento del cuadro en ninguno de los pacientes.
Por el contrario, todos los pacientes con ataxia adquirida a los que no se les realizó tratamiento y casi la totalidad de los de etiología no adquirida tuvieron una progresión del cuadro clínico. Esto no hace más que resaltar la enorme importancia de buscar una causa adquirida en este tipo de pacientes, dado la gran respuesta que se puede obtener al realizar un tratamiento etiológico dirigido. De todas formas, existen casos en los cuales a pesar de realizar un diagnóstico etiológico definido y un correcto tratamiento dirigido, el cuadro continúa progresando (LMP en pacientes severamente inmunodeprimidos, degeneración cerebelosa paraneoplásica y otros).
Conclusiones
1. Más de la mitad de los pacientes con AECA presentaron una etiología adquirida de la ataxia.
2. Las etiologías más frecuentes en este trabajo fueron la degeneración cerebelosa alcohólica y la degeneración cerebelosa paraneoplásica.
3. Las características que se asociaron a ataxia de causa adquirida fueron:
-Presencia de síndrome cerebeloso vermiano puro (noxas tóxicas)
-Asimetría franca del síndrome cerebeloso (LMP)
-Rápida progresión del cuadro
4. Las características que se asociaron a etiologías no adquiridas fueron:
-Síndrome cerebeloso global y homogéneo
-Progresión lenta
-Atrofia de tronco encefálico en la RNM
5. El tratamiento adecuado de las causas adquiridas de ataxia logró detener o mejorar la evolución de un alto porcentaje de pacientes.

