











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
PermalinkIntroducción
El compromiso hematológico es frecuente entre pacientes infectados por el virus de la inmunodeficiencia humana. Múltiples factores entre los que destaca la propia infección, infecciones oportunistas, fármacos y neoplasias determinan anemia, leucopenia con linfopenia y trombocitopemia entre otras manifestaciones. El abordaje clínico de estas situaciones con frecuencia determina el planteo y análisis de múltiples diagnósticos diferenciales.
El síndrome hemofagocítico (SH) o linfohistiocitosis hemofagocítica es una enfermedad infrecuente y grave. De origen autoinmune, se caracteriza por una alteración en la función de las células natural killer (NK) y T citotóxicas con activación macrofágica que lleva a un estado de hiperinflamación y sobreproducción de citocinas.1)(2
Se divide en primaria o genética 3 , y secundaria o reactiva a diversas infecciones, enfermedades autoinmunes, fármacos y neoplasias hematológicas. 4-12
La clínica es inespecífica y la enfermedad se debe sospechar ante fiebre mantenida, hepatoesplenomegalia y citopenias1)(2. El tratamiento se basa en medidas de soporte vital, tratamiento de los factores desencadenantes y supresión de la respuesta inflamatoria descontrolada.4)(13 Se trata de una condición clínica con elevada mortalidad, y en el pronóstico influyen entre otros la etiología subyacente, siendo los casos idiopáticos y secundarios a neoplasias los de peor pronóstico.14)(15
Se presenta un caso de SH en un paciente infectado por el virus de la inmunodeficiencia humana y se realizan consideraciones diagnósticas y terapéuticas a propósito del mismo.
Caso clínico
Se trataba de una paciente de sexo femenino de 24 años, tabaquista, diagnosticada de infección por virus de la inmunodeficiencia humana (VIH) 8 años antes, permaneciendo desde entonces sin tratamiento antiretroviral (TARV).
El año previo a la consulta presentó neumocistosis pulmonar (Pneumocistis jiroveci) por la que recibió tratamiento en base a trimetoprin-sulfametoxazol, iniciándose en ese momento TARV en base a zidovudina, lamivudina y efavirenz. Mostró mala adherencia al TARV, abandonándolo a pocas semanas del inicio. No realizaba controles médicos periódicos por lo que se desconocía carga viral, población linfocitaria y coinfecciones.
Consultó en emergencia por fatigabilidad muscular de un mes de evolución y alteración del estado general. En las 48 horas previas al ingreso agregó fiebre de hasta 38,5°C axilar, tos y expectoración mucosa. Del examen físico destacaba: vigil, mal estado general con desnutrición proteico-calórica (peso 36 kg, talla 162 cm), muguet oral e intensa palidez cutáneomucosa. A nivel cardiovascular presentaba taquicardia de reposo de 130 cpm y presión arterial de 120/70 mm Hg. El examen pleuropulmonar era normal y en la exploración abdominal se encontró una hepatoespelnomegalia regular.
De la analítica destacaba la presencia de anemia severa con hemoglobina de 5.4 g/dL, trombocitopenia con 95000 cél/mm3 y neutropenia profunda con linfopenia (200 glóbulos blancos/mm3, 3 neutófilos/mm3, 7 linfocitos/mm3). La lámina periférica objetivó en la serie roja normocitosis con normocromía, en la serie plaquetaria un recuento normal, y en la serie blanca escasa celularidad con 6% de granulocitos, 14% de monocitos y 80% linfocitos, algunos con aspecto activado.
Se realizó mielograma que evidenció la presencia de un fenómeno hemofagocítico (Figura 1) con sector eritroide y granulocítico escasamente representado.
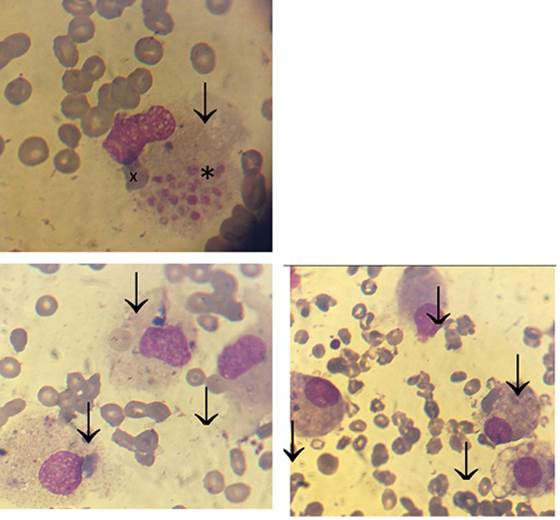
Figura 1: Fenómeno hemofagocítico. Mielograma con tinción May-Grünwald Giemsa (100x). Se observa una médula ósea de muy escasa celularidad con aumento del número de histiocitos/macrófagos y frecuentes fenómenos de hemofagocitosis. En la figura a: se objetiva una célula de gran tamaño (flecha) (histiocito/ macrófago) con citoplasma laxo que engloba un acúmulo plaquetario (*) y un glóbulo rojo (X). En las figuras b y c se observan numerosos histiocitos (flecha) con células sanguíneas en el interior de su citoplasma.
Destacaba del resto de la analítica la presencia de hiponatremia de 129 mEq/L, ferritina mayor a 15000 ng/mL, triglicéridos de 388 mg/dL y fibrinógeno de 258 mg/dL. La velocidad de sedimentación globular (VES) estaba acelerada a 127mm y el valor de proteína C reactiva (PCR) elevada de 297 mg/L. Del hepatograma destacaba elevación de fosfatasa alcalina 558UI/L, de lactato deshidrogenasa (LDH) 479 UI/L y de gamma-glutamil transpeptidasa (GGT): 529 UI/L.
Del estudio de poblaciones linfocitarias destacaba un conteo de CD4+ de 10/mm3 y presentaba una carga viral > 600.000 copias/mm3.
En la tomografía de tórax presentaba micronódulos centrolobulilllares en lóbulo medio y ambos lóbulos inferiores con patrón de árbol en brote. Hemocultivos, urocultivo y cultivo del lavado bronco-alveolar (LBA) no mostraron desarrollo. La búsqueda de tuberculosis en el LBA mediante baciloscopías y reacción en cadena de polimerasa fue negativa. Se solicitó QuantiFERÓN que también fue negativo. La serología para virus hepatitis A, B y C, citomegalovirus, VDRL, toxoplasma Gondii y Aspergillus no mostraron reactividad. Se solicitó serología para virus de Epstein Barr que mostró positividad para IgG siendo IgM negativo.
Para el tratamiento del síndrome hemofagocítico se inició corticoterapia en base a dexametasona 16 mg i/v día, transfusión de glóbulos rojos y factor estimulante de colonias de granulocitos. Dado el estado de inmunodepresiòn severa en este contexto de neutropenia febril se inició cobertura antibiótica en base a meropenem.
Identificadas las lesiones pulmonares descritas y dada la gravedad de la paciente se inició tratamiento dirigido a cubrir Pneumocistis jiroveci (trimetoprin-sulfametoxazol), Mycobacterium tuberculosis (isoniazida, pirazinamida, rifampicina, etambutol y estreptomicina) y micobacteriosis atípica (claritromicina).
Evolucionó favorablemente con el tratamiento instituído, normalizando progresivamente los parámetros clínicos y analíticos. Al momento del alta se reinició TARV en base a tenofovir, lamivudina y nevirapina.
Discusión
Se presentó el caso de una paciente infectada por el VIH con inmunodepresión severa, que desarrolló un síndrome hemofagocítico (SH). La hemofagocitosis es un proceso fisiológico caracterizado por la presencia de eritrocitos, plaquetas o leucocitos en el citoplasma de los macrófagos 1
Aunque es marcador de activación macrofágica y puede formar parte de un SH, el hallazgo de hemofagocitosis aislada no es patognomónico del mismo12.
El SH se define como una estimulación masiva e inefectiva del sistema inmune. Esta estimulación determina aumento de mediadores inflamatorios circulantes e infiltración macrofágica tisular, fenómenos responsables de las manifestaciones clínicas más dominantes 1 Si bien es una manifestación inhabitual en pacientes infectados por el VIH, se ha hallado el fenómeno de hemofagocitosis hasta en 20% de autopsias de pacientes en etapa SIDA 16 Esto sugiere que pueda ser una manifestación subdiagnosticada vinculada a la enfermedad, tratamientos administrados o infecciones oportunistas entre otras.
La fiebre mantenida, presente desde el inicio del cuadro en el caso analizado, usualmente responde a niveles elevados de interleucina 1 e interleucina 6 17)(18. La hepatoesplenomegalia y la elevación de enzimas hepáticas, también presentes en la paciente, son habitualmente consecuencia de la infiltración tisular por linfocitos y macrófagos 14. Las citopenias presentes en el caso analizado, son consecuencia del incremento de factor de necrosis tumoral alfa (TNF-a)2)(14). Este mediador inflamatorio también es responsable de la inhibición de la lipasa proteica, determinando la hipertrigliceridemia objetivada en el lipidograma de la paciente 1).
La ferritina plasmática y los linfocitos CD25+ se comportan como reactantes de fase aguda e incrementan sus niveles como respuesta al estado inflamatorio sistémico. Junto con otros reactantes de fase aguda como la beta 2 microglobulina son marcadores de la severidad de la enfermedad 19).
Se han establecido criterios diagnósticos propuestos por la “Study Group of the Histiocyte Society” revisados en el año 2004 y 2007, que se expresan en la (Figura 2)20). El caso analizado presentaba al menos 5 de los criterios propuestos, entre los que se hallaba la confirmación del fenómeno de hemofagocitosis en médula ósea (Figura 1).

El SH puede vincularse a alteraciones genéticas que determinan inadecuada activación macrofágica. Cuando ello ocurre la enfermedad se manifiesta usualmente en la infancia y es denominado SH primario 3)(21).
Como contrapartida, el SH puede ser gatillado por infecciones, exposición a fármacos, enfermedades autoinmunes y neoplasias 1), denominándose secundario cuando acontece en este escenario. Las infecciones virales son el desencadenante más frecuente, y dentro de éstas las ocasionadas por virus de la familia herpes son responsables de más de 50% de los casos 5)(6)(22-24). Las infecciones bacterianas también han sido identificadas como desencadenante de SH, dentro de éstas la tuberculosis se ha responsabilizado de hasta el 10% de los casos, principalmente en pacientes con inmunocompromiso 12)(25)(26. En pacientes infectados por el VIH el SH puede ser gatillado por infecciones oportunistas, destacando por su importancia la infección tuberculosa 12)(25 y la histoplasmosis diseminada27-29).
Descartadas las infecciones oportunistas, la infección por el VIH puede gatillar un SH30-32, si bien en este escenario ello constituye un diagnóstico de exclusión.
Lo expuesto explica la conducta tomada con el caso descrito, donde se realizó una búsqueda exhaustiva de infecciones oportunistas y frente al hallazgo de imágenes pulmonares patológicas y sugestivas se realizó tratamiento antituberculoso empírico. Por último, la TARV puede gatillar un SH 32 independientemente del tiempo de inicio de la misma una vez que éste se manifiesta.
El esquema terapéutico usualmente incluye el tratamiento de la o las causas que gatillaron el SH, a lo que se asocia la utilización de corticoides a altas dosis para suprimir la respuesta inflamatoria sistémica2)(20). En pacientes con enfermedad aguda severa o que presentan rápido deterioro del estado general, se asocia a los corticoides (usualmente dexamentasona), etopósido 20). En pacientes estables, que no se muestran severamente enfermos, puede tratarse la causa desencadenante (infección, neoplasia, fármacos) y mantener una conducta expectante sin necesidad de administrar quimioterapia de inicio 20). El uso de inmunoglobulina intravenosa se reserva para situaciones en que el SH es gatillado por enfermedades infecciosas (33).
En el caso presentado se realizó tratamiento empírico dirigido a la cobertura de Pneumocitis jiroveci, Mycobacterium tuberculosis y micobacteriosis atípica. Se asoció dexametasona a altas dosis para suprimir la respuesta inflamatoria.
La mortalidad es elevada (20%-80%), con una variabilidad usualmente asociada al desencadenante del SH. En el caso de SH asociado a infecciones puede llegar a 20%-40% y cuando se vincula neoplasias (fundamentalmente hematológicas), la mortalidad puede ser próxima al 100% 12).
Dado lo relevante de su diagnóstico y tratamiento oportunos, el SH debe ser considerado cuando se valora un paciente con infección por VIH y citopenias hematológicas. Algunas claves clínicas son la presencia de marcadores inflamatorios persistentemente elevados (ferritina, VES, proteína C reactiva), persistencia de fiebre a pesar de un tratamiento adecuado de las coinfecciones y esplenomegalia mantenida. El tratamiento debe incluir el tratamiento precoz de las infecciones oportunistas, el inicio temprano de la TARV y el bloqueo de la respuesta inflamatoria sistémica.

