










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Uruguaya de Medicina Interna
versión impresa ISSN 2393-6797versión On-line ISSN 2393-6797
Rev. Urug. Med. Int. vol.1 no.2 Montevideo ago. 2016
Caso clínico
SÍNDROME DE MELAS COMO CAUSA INHABITUAL DE ACV ISQUÉMICO
Presentación de un caso clínico
MELAS SYNDROME as unusual ischemic stroke CAUSE
Presentation of a clinical case
Dra. Mariana Legnani
Residente de Neurología
Dra. Beatriz Arciere
Neurólogo. Servicio de Neurología del Hospital Maciel
Dr. Andrés Boero
Neurólogo. Servicio de Neurología del Hospital Maciel
Dra. Tammara Méndez
Residente de Neurología
Dra. Cristina Pérez
Neurólogo. Jefe del Servicio de Neurología del Hospital Maciel
Recibido: 21/5/16 - Aceptado: 11/7/16
Departamento e Institución responsables: Servicio de Neurología. Hospital Maciel. Montevideo – Uruguay.
Correspondencia: Beatriz Arciere. Dirección: 25 de mayo 172. Servicio de Neurología. Hospital Maciel. CP 11000. Montevideo, Uruguay. Correo electrónico: barciere@hotmail.com
RESUMEN
Las mitocondriopatías son un amplio espectro de patologías caracterizadas por un metabolismo mitocondrial anómalo. Dentro de estas enfermedades se incluye la MELAS, cuya sigla en inglés significa encefalomiopatía mitocondrial, acidosis láctica y episodios “stroke-like”. Se trata de un síndrome de presentación clínica heterogénea que debe considerarse como causa inhabitual de accidente cerebro vascular-símil en los pacientes jóvenes. La resonancia magnética puede otorgar la clave diagnóstica al mostrar áreas de isquemia cerebral sin respeto de los territorios vasculares arteriales. La biopsia muscular con el hallazgo de las fibras rojas rasgadas puede sugerir enfermedad mitocondrial. El diagnóstico definitivo de MELAS se realiza mediante análisis genético por demostración de mutaciones en el ADNm. Se presenta el primer caso clínico confirmado de MELAS con episodios “stroke-like” en la edad adulta reportado en nuestro país.
Palabras clave: MELAS, ACV-símil
ABSTRACT
Mitochondriopathies are a wide spectrum of diseases characterized by abnormal mitochondrial metabolism. Within these diseases is the MELAS, whose acronym means mitochondrial encephalomyopathy, lactic acidosis and "stroke-like" episodes. It is a syndrome of heterogeneous clinical presentation that should be considered as an unusual cause of stroke in young patients. Brain MRI can provide the diagnostic key with areas of ischemia without respect for arterial vascular territories. Muscle biopsy may suggest mitochondrial disease with the finding of ragged red fibers. The definitive diagnosis of MELAS is done through genetic analysis with mutations in the mtDNA. We described the first clinical confirmed case of MELAS with "stroke-like" episodes in adulthood reported in our country.
Keywords: MELAS, stroke-like
Introducción
Las mitocondriopatías son un amplio espectro de patologías que afectan la vía final de la fosforilación oxidativa en la cadena respiratoria mitocondrial. Las mismas pueden deberse a mutaciones en el ADN mitocondrial (ADNm) o en el ADNnuclear (ADNn), sabiendo que el ADNm humano codifica sólo 13 de las 89 subunidades de la cadena. (1,2,3)
La herencia del ADNm es por vía materna en prácticamente todos los casos. Para que se manifieste la disfunción mitocondrial en uno o más tejidos u órganos, existe un efecto umbral, lo que implica que se necesita un número mínimo de moléculas de ADNm mutadas. Asimismo hay heteroplasmia (mitocondrias con ADNm con y sin mutaciones en la misma célula) así como segregación mitótica (las diferentes células y tejidos presentan proporciones variables del ADNm mutado). Esto explica las diferentes presentaciones clínicas fenotípicas, que pueden manifestarse en distintas etapas de la vida. Los tejidos con mayor metabolismo oxidativo y por lo tanto, más susceptibles de ser afectados son: cerebro, ojo, miocardio y músculo esquelético.(1-3)
Dentro de las enfermedades mitocondriales por defectos en el ADNm está lo que se conoce como MELAS, cuya sigla en inglés significa encefalomiopatía mitocondrial, acidosis láctica y episodios “stroke-like” (1).
Es un síndrome de presentación clínica muy variada, que debe ser considerado como causa inhabitual de ataque cerebrovascular-símil en los jóvenes.
Caso clínico
Se presenta el caso de una paciente de sexo femenino, de 32 años, con antecedentes familiares de madre con retardo mental y hermano con patología psiquiátrica y diabetes. Como antecedentes personales se destacan dificultades de aprendizaje e independencia parcial en las actividades de vida diaria, diabetes mellitus insulino requiriente y óbito fetal a las 30 semanas de gestación.
Ingresa a la emergencia del Hospital Maciel por crisis epiléptica focal motora con clonias de hemicara y miembro superior izquierdo con compromiso posterior de conciencia, que reitera en 2 oportunidades. Cefalea frontal intensa de 48 horas de evolución, fiebre de hasta 38°C axilar, dolor abdominal, vómitos y diarrea, agregando en las últimas 24 hs compromiso de la vigilia.
Del examen físico al ingreso se destaca obnubilación leve, dismorfia facial, sin elementos de inmunosupresión clínica, con examen neurológico sin focalidad ni rigidez de nuca, pero con afectación miopática proximal de 4 miembros.
Con el planteo de encefalitis infecciosa en primer lugar y de accidente cerebro vascular (ACV) febril en segundo, se realiza una tomografía (TC) cráneo donde se aprecia un área hipodensatémporo-parietal derecha y calcificaciones gangliobasales (figura 1).
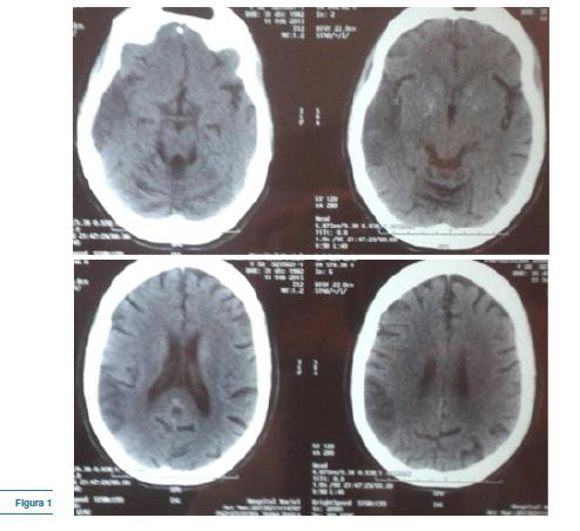
El citoquímico de líquido céfalo-raquídeo (LCR) obtenido por punción lumbar fue límpido, cristal de roca, con una glucorraquia de 1,42 mg/dL para una glicemia de 2 mg/dL, una proteinorraquia ligeramente elevada de 0,52 mg/dL con 2 glóbulos blancos y 2 eritrocitos/mm3. El bacteriológico fue sin desarrollo, así como los hemocultivos. La detección molecular de familia herpes virus y enterovirus en LCR fue negativa.
Las demás rutinas sanguíneas fueron normales y la serología para VIH fue no reactiva. La resonancia magnética (RM) encefálica muestra un área cortico-subcortical temporo-parietal derecha isointensa en T1, hiperintensa en T2 y FLAIR, con restricción en difusión, compatible con área de isquemia aguda del territorio superficial de la ACM derecha. Se destaca el signo del pulvinar bilateral. No hubo alteraciones en el sector venoso ni arterial (figura 2).
El ecocardiograma transesofágico mostró una hipertrofia ventricular izquierda severa con un septum de 17 mm, sin otras alteraciones.
Ante la sospecha de un ACV en el jóven se continuó la valoración etiopatogénica, con ecodoppler de vasos de cuello que fue normal y un holter que mostró preexcitación, compatible con un Wolff Parkinson White (WPW). El VDRL fue no reactivo, así como los ANA y ANCA negativos. Se estudiaron los factores protrombóticos congénitos y adquiridos, destacándose los anticuerpos antifosfolipídicos, todos los cuales fueron negativos.
La paciente presenta una mala evolución posterior, agregando un estado de mal epiléptico que requiere ventilación mecánica invasiva e ingreso a cuidados intensivos. De las gasometrías arteriales se destaca un aumento mantenido del lactato en ausencia de infección generalizada u otras causas.
En sala de medicina agrega episodios de heteroagresividad y trastorno del campo visual compatible con hemianopsia homónima derecha. Con planteo de nuevo ACV se solicita nueva neuro-imagen que muestra un área cortico-subcortical a nivel temporo-parieto-occipital izquierdo, caracterizada por aumento de la señal en T2 y FLAIR con restricción en la difusión, compatible con nueva lesión isquémica que esta vez, no respeta territorio arterial alguno (figura 3).
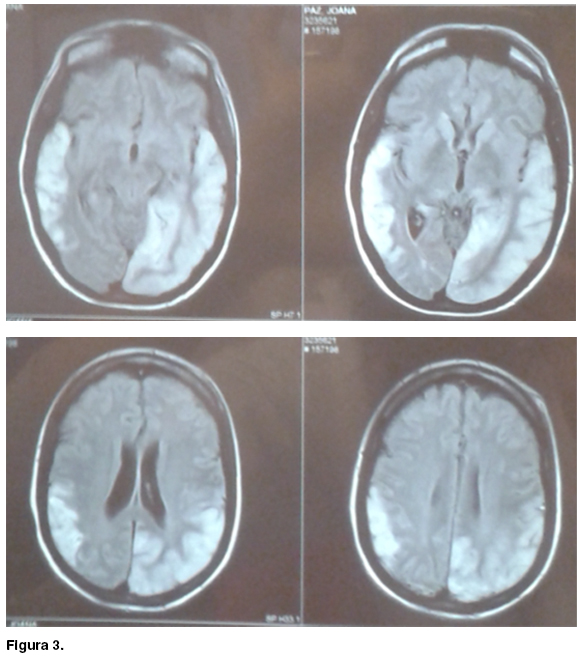
Ante la sospecha de MELAS se realizó el análisis genético por PCR del gen MT-TL1 que mostró la presencia de la mutación 3243A>G en heterocigosis, la cual confirma el diagnóstico planteado.
Discusión
Hemos presentado una paciente que presenta un síndrome de MELAS, en el cual destacan la encefalomiopatía, los episodios stroke-like y la acidosis láctica mantenida, pero también la evidencia de afectación de otros tejidos como analizaremos. Se trata de una enfermedad de herencia materna en la mayoría de los casos, pudiendo estar los familiaresoligo o asintomáticos. Es infrecuente encontrar más de un síndrome de MELAS completo en una misma familia.(2) La paciente analizada tenía una madre con retardo mental y un hermano con diabetes mellitus, lo cual podría vincularse a este tipo de mitocondriopatía.
El debut clínico de la MELAS ocurre a una edad promedio de 10 años, por lo que la mayoría de los casos se diagnostican en la infancia y adolescencia, pudiendo ir de los 2 a los 40 años según se refiere en la literatura. Suelen presentarse con episodios de encefalopatía aguda, cefalea de tipo migrañoso, deterioro de la conciencia y crisis epilépticas, como ocurrió en el caso clínico mencionado. Generalmente existe compromiso cognitivoal momento del diagnóstico ya sea retardo mental o demencia (1,2) que también se evidenció.
Los eventos focales de tipo ACV debutan característicamente antes de los 15 años y usualmente antes de los 40 años (2). Como se aprecia en la neuroimagen de la paciente, los mismos no respetan territorios vasculares arteriales y predominan en la topografía parieto-occipital. (1,2) El hallazgo de calcificaciones gangliobasales también es característico, así como el signo del pulvinar bilateral, el cual además de la MELAS, también puede verse en la enfermedad de Fabry, el sindrome de Wernicke-Korsakoff y la enfermedad de Creutzfeldt-Jakob. (4)
Otras posibles afectaciones son: la miopatía proximal de cuatro miembros simétrica leve-moderada, como la que presentaba la paciente; que a veces asocia compromiso facial, intolerancia al ejercicio, ptosis ocular, oftalmoplejia externa e incluso una polineuropatía sensitiva distal de tipo axonal, donde también puede intervenir como factor etiológico concomitante la diabetes mellitus, frecuente en estos pacientes. (1,2)
Siguiendo con el sistema nervioso central, también pueden observarse elementos extrapiramidales como corea y parkinsonismo, ataxia y sordera neurosensorial. A nivel ocular, retinopatía pigmentaria hasta en el 40%, así como maculopatía y atrofia del nervio óptico (2); elementos no encontrados en este caso clínico.
Respecto al compromiso miocárdico se confirmo en el caso presentado, no sólo una cardiomiopatía hipertrófica, sino también un trastorno de la conducción miocárdica con un WPW, ambos hallazgos compatibles con MELAS.(1,2)
En lo gastrointestinal son frecuentes los trastronos de la motilidad, con alternancia constipación-diarrea, e incluso pseudo-obstrucción intestinal así como vómitos, cíclicos o esporádicos (2); algunos de los cuales la paciente presentó al momento de la admisión en emergencia.
Puede ocurrir fallo renal por diversas circunstancias. También son frecuentes las patologías endocrinológicas con hipopituitarismo, hipotiroidismo, enfermedad de Addison, fallo ovárico y complicaciones obstétricas. (2)
Por diversas razones no se pudo obtener una biopsia muscular, la cual podría mostrar las“ragged red fibers” o fibras rojas rasgadas, no específicas pero sí sugestivas de enfermedad mitocondrial.
El diagnóstico absoluto de esta patología es genético y el mismo está disponible en nuestro país. En este caso, el análisis genético por PCR del gen MT-TL1 mostró la presencia de la mutación 3243 A>G en heterocigosis. Ésta es la mutación más frecuente encontrada en la MELAS, siendo responsable de hasta un 80% de los casos (5), como se reporta en la bibliografía consultada.
En nuestro medio se han presentado casos de pacientes en edad pediátrica con síndromes “stroke-like”, encontrando a la MELAS dentro de las causas etiopatogénicas.(6) No se encontraron, sin embargo, reportes nacionales de casos clínicos confirmados en adultos.
Si bien no existe un tratamiento específico para el síndrome de MELAS, se pueden mencionar algunas medidas para llevar a cabo. Dentro de las drogas antiepilépticas que puedan necesitarse, no se recomienda el uso de valproato de sodio dado que puede producir déficit de carnitina y de esta manera interferir con el metabolismo mitocondrial. Se ha reportado beneficioso el uso de L-arginina tanto para mejorar los síntomas durante los episodios stroke-like, como para disminuir la recurrencia de los mismos. Se recomienda el consejo genético para los individuos afectados y sus familias. (1,2)
Por lo tanto, si bien estamos ante un síndrome de MELAS muy expresivo clínicamente, debemos recordar que también puede presentarse en forma oligosintomática. Es importante tenerlo en cuenta como causa inhabitual de ACV isquémico, sobre todo en jóvenes que se presenten con imágenes de isquemia cerebral que no respeta territorios vasculares.
Bibliografía
1-.Turner C, Schapira A. Mitochondrial Disorders. In: Daroff RB,Fenichel GM, JJankovic J, Mazziotta JC. Bradley´s Neurology in Clinical Practice. 6th ed. Oxford: Elsevier; 2012. p. 1473-1487.
2- Neuromuscular Disease Center (en linea) St. Louis: Washington University; 2016 (acceso 23/06/2016).Disponible en:http://neuromuscular.wustl.edu/
3- Illa L, Bárcena J, Zarranz J. Enfermedades musculares y de la unión neuromuscular. Neurología de Zarranz. 5ta. ed. Barcelona: Elsevier; 2007. p 707-752.
4- García L, Guglielmo R, Illa L. Síndrome de MELAS: TC y RM como herramienta diagnóstica no invasiva. Rev. argent. radiol. 2010;74: 379-383.
5- MT-TL1: mitochondrially encoded tRNAleucine 1 (UUA/G). Genetics Home Reference (on line). Bethesda (MD): The Library; 2016. Cystic fibrosis; (reviewed 2014 Aug; cited: jun. 2016). Available in: http://www.ncbi.nlm.nih.gov/gtr/genes/4567/
6-González Rabelino G. Ataque cerebral isquémico en la infancia y en la adolescencia (en línea). En: XIV Simposio Internacional Enfermedad Cerebrovascular; 2011 Setiembre 09 (acceso: jun. 2016); Montevideo: Grupo de trabajo en Patología Cerebrovascular del Instituto de Neurología.Disponible en: http://neurologiauruguay.org/home/images//acv%20ni%F1os%20i.neurologia.pdf

