











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
Permalink
Los Síndromes Mielodisplásicos (SMD) constituyen un grupo heterogéneo de hemopatías clonales caracterizadas por hematopoyesis ineficaz, displasia medular, citopenias en sangre periféricas y riesgo variable de progresión a Leucemia Aguda Mieloblástica (LAM)1. Los SMD representan una de las neoplasias hematológicas más frecuentes en personas de edad avanzada, con una edad media de presentación de 70 años2,3. La incidencia es de 3-4/100.000 habitantes, con un incremento según la edad de 7.5/100.000 habitantes en individuos entre 60 y 70 años y de 35.5/100.000 habitantes en mayores de 80 años según datos recabados en los Estados Unidos3. Debido al envejecimiento de la población y la utilización de nuevas herramientas diagnósticas en la evaluación de las citopenias persistentes, como la secuenciación masiva (NGS; next generation sequencing), se está observando un incremento en los casos de SMD en los últimos años.
El diagnóstico de los SMD resulta complejo y se basa principalmente en la presencia de citopenias persistentes, el estudio morfológico y citogenético de médula ósea (MO). La histología, citometría de flujo y biología molecular contribuyen en este proceso brindando resultados complementarios interpretados como co-criterios4.
La historia natural de los SMD es variada y refleja diferencias en la biología de los subtipos. Por ende, el pronóstico también es heterogéneo en función del subtipo y es necesaria la estratificación del riesgo según sistemas de predicción. La sobrevida global va desde casi 9 años en pacientes de bajo riesgo a menos de 1 año en los de alto riesgo. Los pacientes de mayor riesgo y aquellos que fallecen en los primeros dos años del diagnóstico presentan como principal causa de muerte la progresión a LAM3. Para la estratificación pronóstica han sido publicados numerosos sistemas que consideran variables como edad, sexo, morfología, porcentaje de blastos, presencia y/o profundidad de las citopenias, requerimiento transfusional, niveles de ferritina y anormalidades citogenéticas, entre otras. Los más utilizados son el Sistema de predicción de riesgo Internacional (IPSS) (5 y el IPSS revisado (IPSS-R) (6, el basado en la clasificación de la OMS (WPSS) (7 y el propuesto por el MD Anderson Cancer Center8. El incremento en la accesibilidad de la secuenciación masiva (NGS) hace que lentamente se vaya incorporando a los estudios iniciales a realizar a un paciente, tanto al debut como en la evolución, incidiendo en la decisión terapéutica9,10. En los últimos años ciertos hallazgos moleculares han sido propuestos e incorporados en los sistemas de predicción de nueva generación11.
El tratamiento de los SMD está ligado a una adecuada estratificación de riesgo. Los pacientes de riesgo bajo se tratan con factores de crecimiento, soporte transfusional y quelación del hierro. Mientras que, los de riesgo alto requieren fármacos hipometilantes, quimioterapia intensiva y trasplante alogénico de progenitores hematopoyéticos (alo-TPH).
En los últimos años se han publicado recomendaciones actualizadas para guiar el diagnóstico y tratamiento en SMD13,14,15,16,17,18. Las mismas incorporan las nuevas herramientas diagnósticas, los hallazgos moleculares y la disponibilidad de nuevos tratamientos de acuerdo al riesgo, especialmente con terapias diana12. La adherencia a estas recomendaciones y guías clínicas se considera indicador de buena calidad de atención y es objetivo de los servicios de salud19. Sin embargo, la utilización de las nuevas herramientas diagnósticas y terapéuticas, así como la adherencia a las nuevas guías en países de Latinoamérica como Uruguay depende de la accesibilidad, los costos y la educación médica continua de los profesionales.
Uruguay presenta una población de 3.3 millones de habitantes, de los cuales 93% habita a nivel urbano con un 50% en Montevideo, su ciudad capital. El cáncer es la segunda causa de muerte luego de la patología cardiovascular, sin datos acerca de la incidencia y prevalencia de SMD. Ante la ausencia de evidencia de cómo son estudiados y tratados los pacientes con SMD en nuestro país, el objetivo de este trabajo consistió en relevar, a través de una encuesta online, las herramientas con las que cuentan los hematólogos para afrontar la complejidad diagnóstico-terapéutica de estos pacientes.
Materiales y métodos
Se realizaron 2 encuestas en 2016 y 2019. Las mismas se basaron en una encuesta diseñada por la subcomisión de SMD de la Sociedad Argentina de Hematología20, compartida con el Grupo Latinoamericano de Mielodisplasia y adaptadas a Uruguay. Se distribuyeron entre hematólogos de Uruguay socios de la Sociedad de Hematología del Uruguay (SHU) vía correo electrónico. Las encuestas se realizaron mediante cuestionario online en la plataforma survey-monkey (SurveyMonkey® Inc.; Palo Alto, California, USA), material suplementario.
Las preguntas eran cerradas de opción múltiple y semiabiertas que incluían aspectos concernientes a la práctica profesional referente a los SMD. Se evaluó el acceso a métodos diagnósticos, la elección de sistemas pronósticos y estrategias terapéuticas.
La participación fue voluntaria y anónima. Se solicitó el consentimiento informado para participar del estudio y para la publicación de los resultados.
Análisis estadístico
Las respuestas válidas fueron analizadas utilizando métodos de estadística descriptiva e inferencial. Para el análisis de las diferencias entre grupos se utilizó el test estadístico de Chi cuadrado y el test no paramétrico de Mann Whitney para las variables continuas. Se consideró significativo un valor de p<0.05. Para el procesamiento de las variables en estudio, se empleó el paquete estadístico SPSS versión 20.0 (SPSS, Chicago, EE.UU.) y el GraphPad Prism versión 5.0 (GraphPad Software, San Diego, EE.UU.).
Resultados
Se incluyeron en la encuesta de 2016, 39 encuestados y en la de 2019, 32 encuestados. La tasa de respuesta fue del 32.5% y 26.6% teniendo en cuenta que la SHU tiene 120 socios activos.
Todos los encuestados asisten exclusivamente pacientes adultos. Los datos de la población encuestada en 2016 y 2019 se muestran en la Tabla 1. Se destaca que 68.4% y 52.5% de los encuestados en los años 2016 y 2019 respectivamente tienen más de 40 años, y 60.5% y 56.2% tienen más de 10 años de ejercicio profesional.
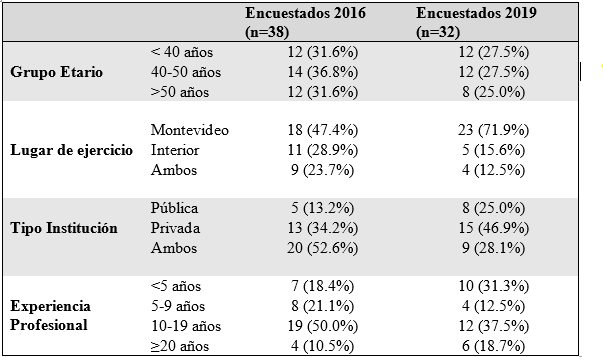
Con respecto a la accesibilidad y aplicación de las herramientas diagnósticas se exploró la histología, citogenética y citometría de flujo (CF). En la encuesta de 2019 se añadió la exploración de citogenética molecular (hibridación in situ con sondas fluorescentes-FISH) y biología molecular. En la Tabla 2 se muestra la disponibilidad de herramientas diagnósticas en cada una de las encuestas.
En relación a la histología, 14 de los 38 encuestados (36.8%) en 2016 refirieron que los estudios histológicos lo realizan hemato-patólogos y el resto un patólogo general. Los estudios histológicos se realizan en el mismo lugar de atención del paciente mayor frecuencia, 78.6% y 75.0% en 2016 y 2019, respectivamente. Los resultados fueron similares para la asistencia pública y privada o interior y capital del país. Este estudio es realizado en más del 75% de los pacientes en según el 100% y 93.8% de los encuestados en 2016 y 2019, respectivamente.
Los estudios citogenéticos y de FISH están disponibles en todos los centros del país, aunque la mayoría requiere derivar la muestra instituciones de Montevideo (Tabla 2). Cuando se interrogó la frecuencia, el estudio citogenético y FISH se realiza en más del 75% de los pacientes en 92.1% y en el 84.4% de los encuestados en 2016 y 2019 respectivamente. No se observaron diferencias estadísticamente significativas entre instituciones públicas y privadas ni entre instituciones de la capital e interior.
Con respecto a la CF, ésta está disponible para realizarse en todos los centros del país, aunque similar al citogenético y FISH, en una elevada frecuencia las muestras deben ser derivadas a otras instituciones en Montevideo (Tabla 2). En la encuesta de 2019 se interrogó a cerca del tipo de citómetro con el cual se trabaja. Trece (40.6%) encuestados desconocen las características, 14 (43.8%) utilizan citómetros de 8 colores o más, 3 (9.4%) de 5 o 6 colores y 3 (9.4%) de 3 o 4 colores. Cuando se interrogó la frecuencia en que sus pacientes se realizan CF, respondieron que se realiza en más del 75% de los pacientes en el 97.4% y en el 93.7% de los encuestados en 2016 y 2019. No se observaron diferencias entre instituciones públicas y privadas ni entre instituciones de la capital e interior del país.
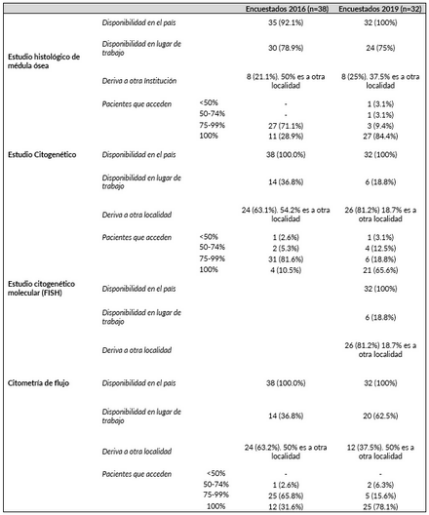
En 2019, 3 de 32 (9.4%) encuestados tienen acceso a paneles de NGS. Todos derivando la muestra al exterior. Sin embargo, algunos encuestados podían realizar estudios moleculares puntuales en nuestro país como: RUNX1 18/32 (56.3%), ASXL1 14/32 (43.8%), IDH1 o IDH2 4/32 (12.5%), SF3B1 8/32 (25%), TP53 24/32 (75%). Al interrogar que porcentaje de sus pacientes eran estudiados con determinaciones moleculares las respuestas fueron: ninguno 2 (6.3%), menos de 25% 7 (21.9%), entre 26-50% 8 (25%), entre 51-75% 2 (6.3%), entre 75-99% 5 (12.5%) y todos 9 (28.1%). Por ende, el 40.6% de los encuestados respondió que más de 75% de sus pacientes son estudiados con determinaciones moleculares. El acceso fue superior en la asistencia pública que en la privada (63% vs 23.5%, p=0.023)
Los hematólogos contestaron, en la encuesta del año 2016, que informan el diagnóstico de SMD a sus pacientes como una falla medular en 86.8% (33) y con menor frecuencia, como estado pre-leucémico 2.6% (1), enfermedad de la sangre 2.6% (1), y cáncer o neoplasia el 7.9% (3). No se observaron diferencias en estos resultados entre instituciones públicas y privadas ni entre instituciones de la capital e interior del país. Esta pregunta no fue incluida en la versión del año 2019.
La mayor parte de los encuestados en el año 2016 utilizan el IPSS (17, 44.7%) o IPSS-R (15, 39.5%) para evaluar el riesgo de la enfermedad. Sólo 3 (7,9%) WPSS, 1 (2.6%) el score del MD Anderson y 2 (5.2%) múltiples sistemas de scores ya que ninguno cumple con las expectativas. No se observaron diferencias en uso de score pronósticos entre instituciones públicas y privadas ni entre instituciones de la capital e interior. Esta pregunta no fue incluida en la versión del año 2019.
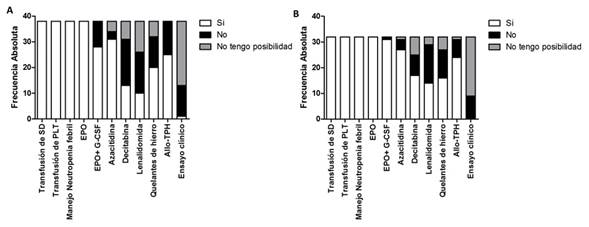
Con respecto a las opciones terapéuticas, en la encuesta de 2016, los médicos informan todas las alternativas terapéuticas a sus pacientes en 13 (34.2%), solo las opciones que considera necesario administrar en 17 (44.7%) y 6 (15.8%) limitan la información a aquéllas que tiene disponibilidad. En la encuesta de 2019, los resultados fueron similares, los médicos informan todas las alternativas terapéuticas a sus pacientes en 14 (43.8%), solo las opciones que considera necesario administrar en 11 (34.4%) y 7 (21.9%) limitan la información a aquéllas que tiene disponibilidad. No se observaron diferencias en estos resultados entre instituciones públicas y privadas ni entre instituciones de la capital e interior del país.
En la Figura 1 se muestran la indicación de los diferentes tratamientos. La indicación de tratamientos entre instituciones públicas y privadas, por grupo etario o experiencia profesional no mostraron diferencias significativas. Al comparar médicos que se desempeñan en Montevideo o el interior no se encontraron diferencias en la mayoría de los tratamientos a excepción de la azacitidina. La indicación de azacitidina fue más frecuente en Montevideo respecto al interior en la encuesta de 2016 (18/18, 100%, en Montevideo y 7/11, 63.6%, en el interior, p=0.04). En 2019 no hubo diferencias estadísticamente significativas entre estos grupos.
Se preguntó sobre cuantos ciclos se esperan a administrar de azacitidina, decitabina y lenalidomida para definir fracaso terapéutico. En 2016, 32 (84.2%) respondieron que administran de 4 a 6 ciclos de azacitidina y 6 (15.8%) 7 o más; mientras que, en 2019, 1 (3.1%) espera menos de 4 ciclos, 24 (75%) de 4 a 6 ciclos y 7 (21.9%) 7 o más ciclos. Con respecto a los ciclos que administran de Decitabina, en 2016, 1 (2.6%) respondió que espera menos de 4, 29 (76.3%) de 4 a 6 ciclos y 8 (21.1%) 7 o más ciclos. En 2019, 9 (28.1%) respondieron que espera menos de 4, 21 (65.6%) de 4 a 6 ciclos y 2 (6.3%) 7 o más. En relación a la Lenalidomida, en 2016, 2 (5.3%) respondieron que esperan menos de 4 ciclos, 24 (63.2%) de 4 a 6 ciclos y 12 (31.6%) 7 o más ciclos. En 2019, 4 (12.5%) respondieron que esperan menos de 4 ciclos, 22 (68.7%) de 4 a 6 ciclos y 6 (18.8%) 7 o más ciclos. No hubo diferencias significativas entre profesionales según edad, experiencia profesional, o que trabajen en sector público o privado.
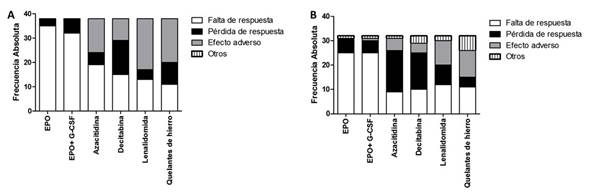
En la Figura 2 se muestran las causas de suspensión de tratamiento en las encuestas de 2016 y 2019. No hubo diferencias significativas en respuestas de los profesionales según edad, experiencia profesional, sector público o privado o interior y capital.
Discusión
Los avances en el diagnóstico y en el tratamiento de los SMD han sido importantes en los últimos años y han cambiado la aproximación clínica de los pacientes con SMD. Realizar un diagnóstico correcto, valoración pronóstica y definir el tratamiento requiere de experiencia debido a la complejidad de esta enfermedad.
El diagnóstico de SMD se realiza según los criterios de la OMS 201621. Se ha descrito que el diagnóstico puede ser establecido ante la presencia de unos “prerrequisitos” junto con al menos uno de los “criterios decisivos o mínimos”. En ausencia de un criterio decisivo, el cumplimiento de los co-criterios puede ayudar a establecer la condición de “sospecha alta de SMD”. Los pre-requisitos son: la presencia de citopenia constante por 6 meses, a excepción de los casos con presencia de exceso de blastos y anomalías citogenéticas relacionadas con SMD, y exclusión de enfermedades hematológicas y no hematológicas como causa de citopenia/displasia. Los criterios mínimos o decisivos son presencia de displasia en al menos el 10% de las células de 1 o más de las líneas mieloides en MO, 5-19% de blastos en MO (o 2-19% en sangre periférica), ≥ 15% sideroblastos, ≥ 5% sideroblastos en anillo en presencia de mutación en SF3B1 y anomalías cromosómicas típicas, por citogenética o FISH. Los co-criterios corresponden a los datos aportados por la histología, citometría de flujo y datos moleculares de clonalidad22. De esto se desprende que contar con citología, citogenética y eventual FISH o biología molecular para la determinación del status de SF3B1 es clave para el adecuado estudio de un paciente con citopenia persistente. Es importante también contar con técnicas complementarias como la histología, la CF y otras determinaciones mediante biología molecular ya que contribuyen, como co-criterios, a dilucidar casos de diagnóstico más difícil.
En este trabajo se demuestra que ya en 2016 existe una amplia disponibilidad y acceso a los estudios de histología, citometría, citogenética y FISH en Uruguay con un importante incremento en 2019. Aunque es necesario derivar la muestra a otra institución o a Montevideo para el caso de pacientes del interior del país no hubo diferencia entre hematólogos que asisten pacientes en interior vs Montevideo ni entre la asistencia pública y privada. Uruguay es un país pequeño donde el envío de las muestras del interior del país a Montevideo lleva solo unas pocas horas. Los resultados de accesibilidad a histología y CF son similares a los reportados en Argentina en una encuesta similar realizada por Crisp et al20. Sin embargo, en Uruguay se observa una mayor accesibilidad a los estudios citogenéticos20,23. En Uruguay la histología el 1/3 de los casos lo realizan hemato-patólogos y en el resto patólogos generales. En la literatura se destaca las dificultades que representa el estudio de un SMD en la histología, así como la discordancia existente interobservador, por lo que la mayoría de los expertos refieren que es importante que sea realizada por patólogos especializados para incrementar su rentabilidad diagnóstica24. Con respecto a la CF se destaca que la disponibilidad en el propio lugar de trabajo se incrementó de 36.8% en 2016 a 62.5% en 2019. Es importante destacar que más de un tercio de los hematólogos desconoce el tipo de citometría que utilizan. En relación a los estudios moleculares, menos de 10% acceden a NGS enviando la muestra al exterior. Sin embargo, el 40.6% de los encuestados refiere que más de 75% de sus pacientes son evaluados con algún estudio molecular y el 25-75% accedía a genes específicos. Se destaca que el 75% refirió acceso a estudios de variantes de TP53 por técnicas de biología molecular tradicionales como secuenciación Sanger. En Uruguay no hay cobertura obligatoria por parte de las instituciones de salud para afrontar los estudios de CF, citogenética, FISH y moleculares por lo que esta financiación varía entre las instituciones privadas, sin costo, en general, en las instituciones públicas. El acceso a la mayoría de las herramientas diagnósticas no se vio afectada entre los sectores públicos y privados a excepción de los estudios moleculares complementarios siendo mayor para pacientes que se asisten en el sector público. Cabe destacar que los estudios moleculares específicos de SMD se realizan, por el momento, únicamente en el Hospital de Clínicas, Universidad de la Repúplica, que forma parte de la red pública de salud.
Los SMD son un grupo heterogéneo de enfermedades con pronóstico variable, en términos de sobrevida global como riesgo de evolución a LAM25. La selección del tratamiento es un desafío frente a la heterogeneidad propia de la patología, la avanzada edad de la mayoría de los pacientes, la presencia frecuente de comorbilidades y elevada morbi-mortalidad de las alternativas terapéuticas con potencial curativo como el trasplante. Por ende, establecer de forma precisa e individualizada el pronóstico de un paciente es esencial para adaptar el tratamiento al riesgo estimado. Existen diferentes sistemas de puntuación que permiten evaluar el riesgo estimado de progresión a LAM y sobrevida. Similar a lo reportado en la literatura y en Argentina, la gran mayoría de los hematólogos encuestados eligen el IPSS o el IPSS-R20,23,26,27,28,29. Estos sistemas incorporan los hallazgos citogenéticos, en conjunto a la presencia de blastos en médula ósea y la profundidad de las citopenias. Diversos grupos han comenzado a publicar sistemas pronósticos que incluyen los datos moleculares y en la actualidad se está trabajando en el desarrollo de un IPSS molecular, cuyos resultados preliminares fueron recientemente presentados en el Congreso de la Americano de Hematología9,10,30,31,32. Por ende, se destaca la importancia de la realización de los estudios citogenéticos y moleculares en la estratificación pronóstica de los pacientes.
La accesibilidad a los tratamientos para SMD es un problema en numerosos países de Latinoamérica, incluyendo Uruguay33,34. Las encuestas exploraron como los hematólogos informan a sus pacientes las opciones terapéuticas. El 30-40% de los encuestados informan a sus pacientes todas las opciones disponibles, alrededor de la mitad sólo las que considera necesario administrar y una minoría limita la información a aquellas que tiene la posibilidad de indicar. Esta conducta relevada está de acuerdo a la tendencia de que los médicos informen y hagan partícipes a los pacientes de las decisiones terapéuticas35. Con respecto a la experiencia e indicación de tratamientos en SMD se observa una amplia disponibilidad de tratamientos de soporte, factores y allo-TPH. La azacitidina, con amplia disponibilidad, incrementó su indicación en el interior del país desde 2016 a 2019 asemejándose a su frecuencia en Montevideo. La disponibilidad o acceso a Decitabina, Lenalidomida y quelantes del hierro era escasa en 2016, con una leve mejoría en 2019. En cuanto al número de ciclos que administran los fármacos antes de definir fracaso terapéutico, la mayoría respondieron 4 a 6 ciclos de azacitidina y decitabina. En cuanto a las causas de suspensión de los tratamientos, los hematólogos indican que la eritropoyetina sola o asociada a G-CSF se suspende por falla de respuesta y la decitabina, azacitidina, lenalidomida y quelantes de hierro lo hace tanto por falta respuesta, pérdida de respuesta o efectos adversos. Estos datos son similares a los reportados en Argentina, aunque la disponibilidad de lenalidomida y quelantes de hierro son mayores en Argentina20,23.
La cobertura financiera de los tratamientos para SMD en Uruguay es insuficiente. Están cubiertos los tratamientos de soporte transfusional, eritropoyetina, aunque ampollas de elevada dosis 10.000 o más unidades puede dificultarse, y factores de crecimiento. En Uruguay existe una entidad pública no estatal que es el Fondo Nacional de Recursos (FNR) que brinda cobertura financiera a procedimientos de medicina altamente especializada, así como a medicamentos de alto costo. Esta cobertura es válida para toda la población residente en el país usuaria del Sistema Nacional Integrado de Salud. En este marco y desde 2018 se otorga cobertura de azacitidina para pacientes de riesgo intermedio y alto hasta 6 ciclos si hay fallo terapéutico o hasta progresión, que antes debían acceder mediante recursos de amparo a través de juicios al Ministerio de Salud Pública. El cambio en la cobertura puede haber influido en el aumento de la indicación en el interior de 2016 a 2019. El FNR no brinda apoyo para Decitabina, Lenalidomida ni quelantes de hierro. Para acceder a estos últimos, los pacientes o la institución deben costear el tratamiento o tramitar acciones legales. La escasa disponibilidad a quelantes de hierro es preocupante. Aproximadamente 80-90% de los pacientes con SMD se presentan con anemia al diagnóstico y un gran porcentaje de ellos se ven expuestos a transfusiones crónicas. Tanto las transfusiones crónicas como la eritropoyesis ineficaz influyen en la sobrecarga de hierro sistémica y tisular observada frecuentemente en pacientes con SMD36,37. La quelación con desferoxamina o deferasirox se recomienda en pacientes con SMD de bajo riesgo con expectativa de vida razonable que están bajo soporte transfusional periódico o presentan sobrecarga férrica previo a un allo-TPH. Se recomienda iniciarlo precozmente, una vez establecida la dependencia transfusional o con ferritina mayor a 1000 ng/Ml38,39. El estudio Telesto que compara deferasirox vs placebo mostró una reducción del riego de 36.4% en sobrevida libre de eventos en pacientes quelados pero sin diferencias significativas en la sobrevida global39,40. Por su parte, la lenalidomida es el tratamiento de elección en pacientes con deleción 5q con anemia sintomática y dependencia transfusional con baja probabilidad de respuesta a eritropoyetina o que han fracasado a la misma. Hay 2 importantes estudios (fase II y fase III) que muestras que Lenalidomida tiene alrededor de un 60% de respuestas con independencia transfusional y alrededor de un 50% de respuestas citogenéticas42,43. La escasa disponibilidad de Lenalidomida en nuestro país limita el tratamiento de pacientes con SMD con deleción 5q que representan aproximadamente al 15% de los SMD41.
El FNR también brinda cobertura para la realización del alo-TCP que se sostiene como la única estrategia curativa capaz de modificar el curso natural de la enfermedad. Sin embargo, su indicación es limitada debido a la elevada edad. Todos los pacientes eventualmente perderán la respuesta a las restantes opciones terapéuticas, en al caso de haber sido alcanzada, en un corto o mediano plazo y, las guías actuales recomiendan la oferta de ensayos clínicos a estos pacientes. Cabe destacar la ausencia de ensayos clínicos para SMD en Uruguay, común a otras patologías dentro y fuera de la hematología. La investigación clínica es una actividad básica para la generación de mejoras en la atención sanitaria que brinda la posibilidad a los pacientes a acceder a nuevos tratamientos.
En conclusión, en Uruguay existe amplia disponibilidad de herramientas diagnósticas para SMD, sin diferencias en la salud pública y privada, a excepción del acceso a ciertas determonaciones por biología molecular. En cuanto a las herramientas terapéuticas, se dispone de tratamiento de soporte, azacitidina y allo-TPH, con dificultades en el acceso a decitabina, lenalidomida y quelantes de hierro y carencia de ensayos clínicos. Consideramos que contar con estos datos, aportados por un elevado porcentaje de hematólogos del país, permite conocer nuestra realidad y tomar acciones para mejorar y optimizar el diagnóstico y tratamiento de los pacientes con SMD con el objetivo de realizar mejoras sanitarias.

