











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
Obviamente, para comenzar este artículo debemos definir nuestro objeto de estudio, o sea, preguntarnos qué es un genoma. Más allá de distintas definiciones que se pueden proponer, podríamos decir, desde un punto de vista operativo, que un genoma es el conjunto completo del ADN dentro de una célula. Por lo tanto, en eucariotas el genoma es el ADN que se encuentra en el núcleo, en las mitocondrias y, en el caso de hablar de plantas, en los cloroplastos. En este artículo nos referiremos en forma exclusiva al genoma nuclear. Desde una amplia perspectiva, el estudio de la organización y evolución de los distintos genomas (eucariotas, procariotas, virales) resulta de interés para diversas áreas de la biología. Por ejemplo, para los biólogos moleculares es de importancia saber cómo se organiza el material hereditario y cómo se distribuyen las secuencias codificantes (aquellas que son transcriptas a ARN mensajero (ARNm) y luego éste es traducido a proteínas) en los cromosomas; cuál es el complemento total de genes, cuál es su distancia media; el número de intrones, conocer los sitios específicos en que aumenta la tasa de mutación y recombinación; la posible influencia de la composición genómica (frecuencia de bases) para comprender el bandeo y los rearreglos cromosómicos así como la estructura de la cromatina, etc. Desde una perspectiva complementaria, los biólogos moleculares especialistas en genómica intentan disecar las bases moleculares, bioquímicas y biofísicas que puedan subyacer a las características antes mencionadas. Finalmente, aunque no por ello menos importante, los evolucionistas comparan los distintos tipos de organización genómica para tratar de conocer los factores causales que determinaron los cambios, a veces drásticos, que se encuentran entre los distintos niveles de complejidad evolutiva, desde los virus y procariotas hasta los mamíferos y plantas superiores.
Naturalmente, los avances en esta ciencia han provocado una revolución no solamente en las ciencias llamadas “básicas”, sino también en aplicaciones concretas para intentar comprender incluso los sistemas biológicos más complejos, como el cerebro humano. Quizás sea conveniente desde el inicio distinguir la genética de la genómica: mientras que la primera se dedica (en lo esencial) a estudiar genes individuales (o pocos) a fin de comprender su funcionamiento y rol en la herencia de determinados caracteres, la genómica estructural utiliza fundamentalmente la secuenciación del ADN y mediante el uso de herramientas computacionales (la llamada bioinformática) ensambla los fragmentos obtenidos a fin de reproducir su orden en el genoma original, localiza e individualiza los genes y, fundamentalmente, analiza la función, estructura y evolución de los genomas completos.
Para la ciencia de la genómica, ocurrió una gran revolución cuando se desarrollaron las técnicas bioquímicas y de bioinformática que permitieron el secuenciado del primer organismo procariota. Esto ocurrió en 1995 cuando se publicó la secuencia completa y el número y tipo de genes de la bacteria Haemophilus influenzae, que es un organismo de vida libre, con un genoma relativamente pequeño de 1.830.140 pares de bases (pb) y que codifica sólo 1.740 genes1. Es necesario aclarar que a este genoma se lo considera “pequeño” ya que, por ejemplo, el genoma humano, como veremos más adelante, tiene aproximadamente 3x109 pb, distribuidos en 23 cromosomas y codifica para aproximadamente 20.000 o 25.000 genes... o sea, en cifras “redondas”, mientras que el genoma típico de las bacterias tiene un tamaño en el entorno de 1,5x106 pb a aproximadamente 10x106 pb, los mamíferos tenemos genomas de 2 o 3 x 109 pb, lo cual implica que nuestros genomas tienen tres órdenes de magnitud más ADN que el “procariota promedio”. Además, en procariotas suele haber un solo “cromosoma” de permutación circular, mientras que en mamíferos encontramos habitualmente más de 20 cromosomas lineales.
Naturalmente, la publicación de la secuencia genómica completa de H. influenzae constituyó un mojón y marcó un salto cualitativo en lo que se refiere a la genómica. En poco tiempo, otros procariotas fueron secuenciados (Mycoplasma genitalium fue el segundo)... y desde ese momento, el mundo de la genómica ya no sería el mismo. Y el crecimiento de los genomas completos disponibles, con la anotación de sus respectivos genes, ha sido exponencial. En las líneas siguientes nos centraremos en las características generales del genoma humano. Si bien los primeros borradores fueron publicados en el 20012)(30, la cobertura y la exactitud de la secuencia se siguen mejorando permanentemente. En las siguientes líneas haremos una breve revisión de las principales características estructurales y evolutivas del genoma humano.
El genoma de los vertebrados
A principios de la década de 1920, Hans Winkler3 acuñó el término “genoma” para definir a la totalidad de genes (en una célula haploide) de un organismo. Naturalmente, las secuencias no codificantes y de otro tipo, como por ejemplo los transposones (secuencias con la potencialidad de moverse de un sitio a otro del genoma), las secuencias reguladoras de la actividad génica, los pseudogenes (“reliquias” de secuencias que fueron activas en el pasado, pero que perdieron su función y comienzan a acumular mutaciones), los intrones (secuencias que interrumpen la parte codificante de los genes y que no están representados en el ARNm maduro ni, por lo tanto, en la proteína), etc., no eran conocidas en ese momento y por lo tanto no fueron incluidas en la definición.
La diferencia más clara entre los organismos vivos está dada por la ausencia o presencia de un compartimento nuclear definido en el que se encuentra el genoma. Los organismos sin núcleo son llamados colectivamente “procariotas” mientras que el otro grupo está constituido por los “eucariotas”. Las bacterias y las arqueobacterias son procariotas, mientras que el resto de los seres vivos, incluyendo los mamíferos y plantas superiores, somos eucariotas. A nivel de organización genómica también existen diferencias significativas entre ambos tipos de organismos. Por ejemplo, el genoma de los procariotas es en la amplísima mayoría de los casos, una molécula única de ADN de permutación circular, cuya longitud en pares de bases (pb) puede ir de menos de 5x105 (sobre todo en bacterias parásitas intracelulares obligatorias) hasta 1x107, y en la cual los genes se encuentran distribuidos en forma muy compacta, siendo por lo tanto la mayor parte del ADN codificante (transcripto a ARN) o con funciones regulatorias.
En los organismos eucariotas la situación es radicalmente diferente. En primer lugar, el material genético está organizado en moléculas de ADN lineales individuales (que junto con determinadas proteínas constituyen los cromosomas) en las cuales dos genes ligados (situados uno a continuación del otro) se encuentran, en general, separados por distancias del orden de pocos cientos (en eucariotas unicelulares) a varias decenas de miles de pb (en plantas y animales “superiores”). En segundo lugar, la cantidad de ADN por genoma haploide (cantidad de ADN de los gametos) varía desde aproximadamente 2,5x107 pb para eucariotas unicelulares, hasta valores del orden de 1011 pb para algunas plantas y anfibios. A su vez, el número de genes es de aproximadamente 4x102 hasta 104 en procariotas; 5x103 en eucariotas unicelulares y 2,5x104 en mamíferos (ver lista completa en https://www.ncbi.nlm.nih.gov/genome/).
Cuando se considera la gran cantidad de ADN por genoma haploide que caracteriza a los organismos estructuralmente complejos como los vertebrados y plantas superiores, surge inmediatamente el problema de cómo este material genético se organiza. Efectivamente, es posible postular que debe existir algún tipo de “orden” que, entre otras funciones, habilite que los miles de genes de, por ejemplo, un mamífero, se expresen en forma ordenada, tanto desde el punto de vista espacial (entre los distintos tejidos del organismo) como temporal (durante las distintas fases del desarrollo). La magnitud del problema resulta más obvia si consideramos que la totalidad de las secuencias codificantes (o sea, solo los exones) representan, por ejemplo en mamíferos, menos del 1% de todo el ADN nuclear. Algunos otros puntos vinculados con el mismo problema organizativo son:
1) ¿existen diferencias entre las zonas del genoma -y de la cromatina- en las que se ubican los genes housekeeping (en castellano “amas de casa”, que son las secuencias que se transcriben todo el tiempo en todas las células del organismo) y los espacial o temporal específicos?;
2) determinadas características morfológicas de los cromosomas metafásicos, como las bandas, ¿tienen una contrapartida a nivel de la organización genómica?;
3) esta organización genómica ¿es conservada a lo largo de la evolución?, o sea, organismos emparentados filogenéticamente ¿presentan un tipo de organización genómica similar? Estas y otras preguntas intentaremos analizar en los siguientes párrafos.
Organización del genoma en isocoros
Una característica importante y crucial de la organización genómica de todos los mamíferos (incluyendo, por cierto, a humanos) y que ha generado mucha polémica entre los especialistas en el tema, es la presencia de zonas o regiones que difieren significativamente entre sí en la frecuencia relativa de las cuatro bases que constituyen el ADN. Este tipo de organización (que veremos la importancia fisiológica que tiene), fue descubierto hacia mediados de los 70 del siglo pasado por el grupo liderado por Giorgio Bernardi, previo a que se descubriesen las técnicas de secuenciación3)(4)(5)(6)(7. Cuando el ADN genómico nuclear de vertebrados o plantas superiores es centrifugado bajo determinadas condiciones8, las moléculas de ADN se separan de acuerdo a su composición de bases en un número discreto de familias, las que, a su vez, están definidas por diferentes niveles de contenido en GC (contenido molar de las bases guanina + citosina). Estos segmentos fueron denominados “isocoros”, o sea, “regiones iguales”. El nombre se debe a su característica fundamental, o sea, que dentro de un isocoro la composición de bases, definida como contenido en GC, varía relativamente poco.
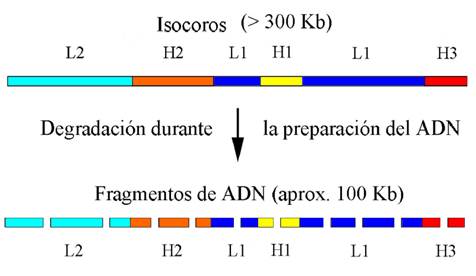
Figura 1: Esquema de la organización en isocoros del genoma humano. Se aprecia la estructura en “mosaico”, en el que los isocoros se alternan sin un orden específico. Durante la preparación para su análisis en la centrífuga, se degradan por acción mecánica, a fragmentos de aproximadamente 100 kb
En la Figura 1 se muestra cómo fueron descubiertas estas estructuras. Dado el tamaño gigantesco de cada molécula de ADN de cada cromosoma, es imposible aislarlas para su posterior análisis (en este caso ultracentrifugación) sin que se rompan al azar. El estudio de los perfiles obtenidos en la ultracentrífuga evidenció que la estructura más probable es la que se muestra en la figura, que podemos resumir en:
a) el genoma es composicionalmente heterogéneo (lo cual contrariaba la opinión dominante a mediados de los ´70s, cuando fueron hechos estos descubrimientos),
b) hay determinados isocoros (definidos por su contenido en GC%) más frecuentes que otros,
c) los isocoros miden mínimamente 100.000 pb (o sea, 100 kb) y la transición entre ellos es relativamente brusca,
d) existen cinco familias de isocoros: dos de ellas “pobres” en GC (L1 y L2), y tres con un contenido más elevado en estas bases (H1, H2 y H3). Desde el punto de vista de su contenido relativo, las familias L constituyen (juntas) el 63% del genoma, mientras que las H son el 24,3%, 7,5% y 4,7%, respectivamente (Figura 2a);
e) las distintas familias de isocoros se hallan alternadas sin un orden específico (Figura 1), por lo tanto los genomas de los vertebrados (particularmente de mamíferos y aves) están formados por un verdadero mosaico de isocoros (ver más adelante).
Fuente: elaboración propia
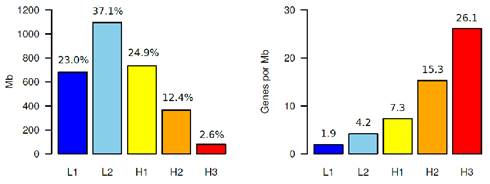
Figura 2 A la izquierda se muestra el perfil composicional de las familias de isocoros del genoma humano. Cada barra representa las cantidades relativas (en %) de los componentes principales respecto al total del genoma. A la derecha se grafica el número de genes por Mb en cada una de las familias de isocoros
Este “perfil composicional” del genoma humano es prácticamente idéntico al de la mayor parte de los mamíferos, lo que sugiere que el contenido en GC de los isocoros puede ser el resultado (y estar sometido) a la acción de la selección natural. Es importante tener en cuenta qué significa “familia" de isocoros. Cuando se afirma que en el genoma humano hay cinco familias de isocoros, no se debe entender que los únicos valores posibles son los que definen a L1, L2, H1, H2 y H3, sino que en realidad estos valores son los más representativos (medios) de cada familia, y el resto se distribuye en forma normal alrededor de cada valor medio9)(4)(5)(10)(6.
Fuente: elaboración propia
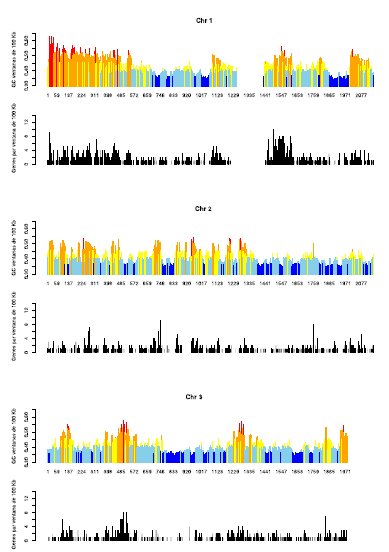
Figura 3: se muestra la distribución de las familias de isocoros (en color) de los cromosomas humanos 1, 2 y 3, indicando el contenido en GC (ordenadas) y la posición, medida en Mb (abscisas). Las zonas “en blanco” constituyen o regiones con muy baja calidad de secuencia, o los centrómeros. Los cromosomas están dibujados en proporción a su longitud. Debajo de cada uno de ellos (en negro), se muestra (en la misma escala) dónde están ubicados los genes en cada cromosoma.
En la Figura 3 se muestra el patrón de isocoros (parte coloreada) de los cromosomas 1, 2 y 3 del genoma humano, y debajo de cada uno de ellos, la ubicación de los genes en los mismos (en negro). Es importante destacar que para los demás cromosomas (datos no mostrados), el patrón es esencialmente el mismo. Para visualizar la organización en isocoros se “cortó” cada cromosoma en fragmentos no solapantes de 100 kb, y a cada fragmento así obtenido se le calculó el contenido en GC. Luego, cada barra fue coloreada de acuerdo a la siguiente clave: GC<37%, color azul (isocoros L1); entre 37% y 41%, celeste (isocoros L2); entre 41% y 46%, amarillo (isocoros H1); entre 46% y 53%, anaranjado (isocoros H2), y finalmente GC%>53, rojo (isocoros H3). Varias conclusiones se pueden extraer de esta figura. La primera, y más obvia, es que los métodos computacionales directos, o sea, la observación fragmento por fragmento, confirman que los cromosomas humanos son un mosaico de isocoros. Segundo, la mayor parte de los cromosomas (sobre todo de los más largos) está conformado mayoritariamente por isocoros L1 y L2. Tercero, los isocoros más ricos en GC (H2 y H3, anaranjados y rojos) tienden a estar ubicados hacia los telómeros (extremos) de los cromosomas. Cuarto, el patrón de distribución de los isocoros es cromosoma-específico, aunque existe una tendencia a que, en promedio, los cromosomas más pequeños tengan más isocoros ricos en GC.
Consecuencias de la organización en isocoros
El descubrimiento en sí mismo de que el genoma de los mamíferos es composicionalmente discontinuo, constituyó una sorpresa. Efectivamente, asumiendo que nuestro genoma seguía “las reglas” del genoma de los procariotas, se pensaba que, al igual que sucede con las bacterias, el genoma “mamífero” iba a oscilar muy poco alrededor de su valor medio (que es aproximadamente 39-40% de GC). Pero a medida que se profundizaba en el tema, se comenzó a comprender que esta heterogeneidad composicional estaba asociada a diversas características11)(3.
a) Distribución de genes
En principio, se podía asumir una hipótesis muy razonable. Asumiendo, como muestra la Figura 2a, que la frecuencia de cada familia de isocoros es diferente, siendo, como dijimos más arriba, las pobres en GC (L1 y L2) el 60% del genoma, entonces resultaba lógico pensar que el 60% de las secuencias génicas (que codifican para proteínas) iban a estar ubicadas en L1 y L2. Sin embargo, esto no solo no es cierto sino que, en realidad, sucede precisamente lo contrario: hay más genes cuánto más ricos en GC son los isocoros, llegando a ser la relación aproximadamente entre H3 (2,6% del genoma) y L1+L2 (60% del genoma) aproximadamente 4,3 (ver Figura 2b y la ubicación de los genes por cromosoma en la Figura 3).
O sea, dónde hay menos ADN (isocoros ricos en GC, familias H1, H2 y H3) hay más secuencias génicas. Este resultado fue del todo inesperado, ya que implica que, de alguna manera, los genes ubicados en las familias de isocoros H (sobre todo en H3) están “más apretados” que los que están en las familias L y, fundamentalmente, la distribución de genes es no aleatoria y dependiente del contenido en GC de cada isocoro.
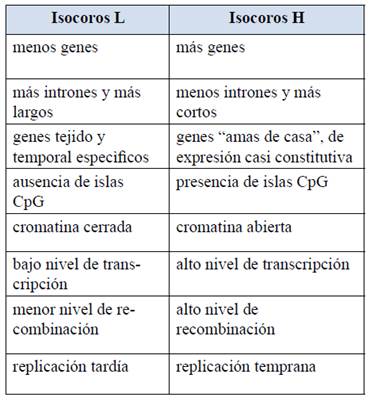
Pero además, y muy importante, hay diferencias significativas entre los genes ubicados en los isocoros L y H (Tabla 1). Pasamos a discutirlas.
Tabla 1: Algunas propiedades estructurales y funcionales de las familias de isocoros L y H. Como se aprecia, las características fundamentales estructurales y funcionales dependen de la composición del genoma, y son opuestas en las regiones pobres (L) y ricas (H) en GC.
Fuente: elaboración propia
1) La mayoría de los genes situados en las regiones más pobres en GC (que denominaremos genes L), suelen ser genes temporal o espacialmente regulados. Es decir, son mayoritariamente secuencias que se expresan solamente en determinados estadios del desarrollo y/o tejidos específicos. Por el contrario, los ubicados en los isocoros ricos en GC (que denominaremos genes H) tienden a ser secuencias housekeeping, o sea genes que se expresan en todo momento y en la mayoría de los tejidos.
2) Los mecanismos de regulación de los genes L y H difiere: mientras que los primeros suelen tener en sus secuencias reguladoras los llamados “TATA box” (secuencias ricas A+T que se encuentran 5' respecto al inicio de la transcripción, los segundos dependen menos de la presencia de estas secuencias para la regulación de su actividad12.
3) Para explicar este punto conviene discutir brevemente el código genético. Recordemos que existen 64 codones para codificar los 20 aminoácidos que constituyen nuestras proteínas. De ese total, tres significan “fin de lectura”. En definitiva, nos quedan 61 codones para 20 aminoácidos. Esto implica, obviamente, que varios codones van a “significar” durante la traducción el mismo aminoácido: estos son los llamados codones sinónimos. Excepto Metionina (codificada por ATG) y Triptófano (TGG), los 18 aminoácidos restantes son codificados por dos, tres, cuatro o seis codones. Y como regla general, el cambio (entres los sinónimos) ocurre en las terceras posiciones del codón (llamadas, por eso, posiciones sinónimas) y en esa posición los cambios G por A o C por T no alteran el aminoácido. Por eso es que habitualmente se dice que las terceras posiciones tienen un grado de libertad de cambio muy alto (y de hecho son las que más cambian), y por lo tanto se puede alterar radicalmente el contenido en GC3 global de un gen sin cambiar los aminoácidos codificados. Dado que existen correlaciones significativas y positivas entre el contenido en GC3 (o sea, de las terceras posiciones de los codones, que como dijimos son las que más libertad tienen de cambiar sin alterar el aminoácido codificado) y los isocoros donde los genes se encuentran (ver más abajo), el uso de codones sinónimos varía enormemente entre los genes L y los H.
4) A su vez, dado que también a medida que los genes se ubican en isocoros más ricos en GC, también aumenta el GC de las posiciones 1 y 2 de los codones (las que tienen mayor poder codificante), el uso global de aminoácidos también difiere en los genes L y los genes H 13.
b) Patrones (patterns) composicionales
Una forma de estudiar los genomas, es analizar la distribución de acuerdo al contenido en GC de los fragmentos del propio genoma, de los valores de GC3 de los genes, de los intrones, exones, etc. Por ejemplo, el histograma que grafica la distribución composicional de los fragmentos no solapantes de todo el genoma humano junto (mostrado en la Figura 4), representa un patrón composicional que refleja, a su vez, el tipo de isocoros característico del genoma humano.
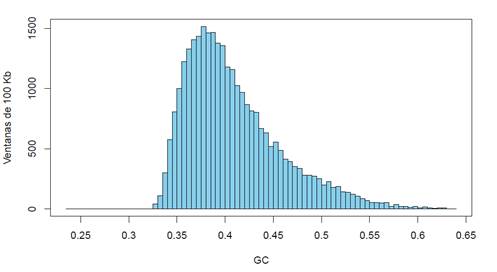
Fuente: elaboración propia
Figura 4: histograma del contenido en GC del genoma humano cortado en fragmentos de 100 kb. En la ordenada se muestra el número de fragmentos (ventanas) para cada contenido en GC (mostrado en la abscisa)
Es importante reiterar que el genoma humano, desde este punto de vista, no difiere significativamente de cualquier otro genoma de mamífero, viéndose en cambio, algunas diferencias notables cuando lo comparamos con otros vertebrados (este punto está por fuera de los objetivos de esta revisión). Si miramos en detalle la Figura 4 podemos extraer varias conclusiones, entre ellas: a) el genoma humano presenta una distribución de fragmentos que van desde aproximadamente un 32% de GC hasta algo más de 60%. b) La distribución, si bien recuerda una campana, en realidad es asimétrica, ya que “sube” rápidamente hacia el valor de la moda (aproximadamente 37% de GC) y baja en forma pausada hacia los valores altos de GC. Naturalmente, esto es una consecuencia de la estructura discutida más arriba en la que se muestran las frecuencias relativas de las distintas familias de isocoros (Figura 2). c) Una observación cuidadosa muestra que el descenso desde la moda hacia los valores máximos de GC no es “suave” sino que existen algunas irregularidades. Por ejemplo, a 42% de GC existe un “hombro”, en 45% de GC se observa una leve subida, entre 48% y 49% la cantidad de fragmentos prácticamente no baja, nuevamente hay un ascenso en 51%, etc. Estas pequeñas alteraciones nuevamente son consecuencia de la estructura en isocoros y de los porcentajes relativos diferentes de ADN de cada familia.
Otros dos tipos de histogramas, también muy ilustrativos, son los que se muestran en la Figura 5.
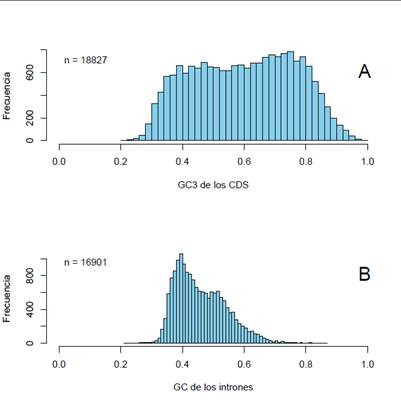
Fuente: elaboración propia
Figura 5: histogramas de A) contenido en GC3 de los 18.827 genes completos analizados en este estudio, B) del contenido en GC de los intrones (16.901), dado que hay un porcentaje menor, pero importante, de genes sin intrones. Por detalles, ver texto
En la parte a) se observa la distribución de los valores de GC3 de los exones humanos disponibles en los bancos de datos. El cálculo es simplemente contar el número de terceras posiciones de los codones que terminan en G o C, y dividir este número por el total de terceras posiciones para cada gen. Nuevamente, hay algunas características interesantes de esta distribución. En primer lugar, la distribución es claramente bimodal, existiendo dos picos: uno centrado en aproximadamente 47% de GC3 y el otro en un valor cercano a 75%. Segundo, esta distribución reafirma el concepto ya establecido de que hay más genes (y más ricos en GC), a pesar de que las familias de isocoros en las que están presentes son menos abundantes. Tercero, el hecho de que haya genes con un contenido en GC3 mayor a 90%, implica necesariamente que, para codificar las proteínas, usan prácticamente la mitad de los codones disponibles. Naturalmente, esta distribución muestra nuevamente que el uso de codones difiere grandemente entre los genes humanos.
En la Figura 5b observamos la distribución composicional de los intrones. Recordemos que los intrones son secuencias que se encuentran en el gen (o sea, en el ADN), que son transcriptas por la ARN polimerasa pero son eliminadas durante el procesamiento del ARN mensajero en el núcleo, por lo cual su secuencia no es traducida. Además, es importante destacar que la mayoría de los genes de mamíferos tienen intrones, aunque no todos, que la longitud de ellos es variable, como también lo es la cantidad de intrones por gen. Por ejemplo, en nuestro genoma los intrones representan aproximadamente el 50% del genoma; su longitud va desde unos 50 pb hasta más de 1.000.000 de pb (lo cual representa el tamaño de un genoma bacteriano pequeño) aunque el promedio es 5.900 pb y el número promedio de intrones por gen es de aproximadamente 10. Finalmente, destacamos que solo alrededor del 10% de los genes no poseen intrones14. La figura fue construía uniendo todos los intrones de cada gen (o sea, “fabricando” artificialmente un intrón único por gen) y haciendo el promedio de GC% para cada uno de ellos. Esta figura también merece algunas consideraciones. Primero, la distribución es bimodal, pero muy sesgada hacia la “izquierda”, o sea, tiene un pico centrado en un valor relativamente bajo, de aproximadamente 38% de GC y luego presenta un segundo, menos importante cuantitativamente, con un valor de GC de 50%. Segundo, a partir de este valor se produce una caída brusca hacia la “derecha” (valores más altos de GC), y tercero, en la práctica, no hay intrones con un GC% mayor a 70. Si asumimos que los intrones, al menos en la mayor parte de su secuencia, son selectivamente neutros, o sea, se pueden producir cambios en la composición de bases sin alterar su funcionamiento y, como dijimos más arriba, asumimos también que el GC% de las terceras posiciones de los codones son también neutros, al comparar los dos histogramas de la Figura 5 vemos que dos componentes “neutros” tienen comportamientos notoriamente diferentes, y en particular, resulta claro que los intrones tienden a presentar valores de GC bajos. Este punto lo veremos en detalle en la próxima sección. Por ahora baste decir que se pueden realizar estudios similares con las posiciones 1 y 2 de los codones, con el uso de aminoácidos y codones de cada familia de isocoros, de dinucleótidos (o de otros oligonucleótidos más largos). Y todas estas características juntas, o sea, estos “patrones” composicionales, definen fenotipos genómicos que, como decíamos más arriba, son similares o idénticos para organismos cercanos filogenéticamente, pero pueden diferir para especies no emparentadas. Es decir, las características composicionales del genoma constituyen, de por sí, un fenotipo. Un hecho importante es que el patrón de los vertebrados de sangre caliente (aves y mamíferos) difiere mucho del patrón de los vertebrados de sangre fría (peces, anfibios y reptiles), lo que se ha vinculado con el origen de los isocoros ricos en GC, característico de los primeros. Esta discusión evolutiva escapa a los objetivos de esta revisión, pero recomendamos leer, entre otros, los siguientes artículos:3)(5)(15)(16)(17)(7.
c) Correlaciones composicionales
Como ha quedado expuesto, el genoma humano -como el de otros mamíferos- es composicionalmente heterogéneo, y este rasgo determina muchas de las características funcionales que definen a nuestros genomas. Y como vimos, los genes obviamente se encuentran inmersos en los isocoros, cada uno de ellos definido por un distinto contenido en GC. Y podemos preguntarnos cómo afecta a las “distintas” regiones de un gen (valor medio de GC en las posiciones 1, 2 y 3 de los codones), o a la composición de los intrones, a la frecuencia de dinucleótidos, etc., la presencia en cada gen. En otras palabras, ¿existe algún tipo de correlación composicional entre los isocoros en los que están ubicados los genes, y la composición global de estos y de sus constituyentes? ¿Si un gen está ubicado, digamos, en un isocoro L, su composición en GC3 y la de sus intrones, será baja; y lo inverso pasará si está ubicado en un isocoro H? Este análisis de correlaciones composicionales comenzó a hacerse en cuanto fueron disponibles un número aceptable de genes humanos y de las secuencias que lo rodeaban, asumiéndose que éstas (aunque no eran al inicio muy largas por las dificultades de secuenciación) eran representativas de los isocoros9)(8)(18. Las conclusiones que se obtuvieron de estos trabajos pioneros, y que hoy están plenamente confirmadas (como mostraremos más adelante con un par de ejemplos), es que existen correlaciones composicionales (o sea, contenido en GC) positivas y estadísticamente significativas entre a) los isocoros y los exones ubicados en ellos, b) entre los isocoros y cada una de las tres posiciones de los codones, siendo más fuerte con la posición 3 (que es la que más puede variar), c) entre los isocoros y los intrones de sus genes, d) entre las tres posiciones de los codones, e) entre las posiciones 3 de los codones y los intrones correspondientes, etc. Estas correlaciones composicionales muestran que a medida que los genes están ubicados en isocoros más ricos en GC, también aumenta el GC de todos los componentes del gen, desde los intrones hasta las tres posiciones de los codones.
Este tipo de correlaciones composicionales (así como otras que también existen pero que no tenemos espacio para discutir)19 son importantes por dos aspectos diferentes. En primer lugar, le dan apoyo a la idea que postula que las fuerzas que determinan la composición de bases de un determinado isocoro operan siempre en la misma dirección, aunque con intensidad variable, sobre todas las secuencias que están en él, independientemente de la función que cumplan. Esto lleva inclusive a que exista una correlación también positiva entre el contenido en GC3 y el contenido en GC de las posiciones 1 y 2 de los codones de cada gen. Esta correlación tiene un importante significado funcional, ya que implica que los genes ubicados en los isocoros más ricos en GC tienden a codificar, con una frecuencia más alta que las secuencias que están en L1 y L2, para el subgrupo de aminoácidos codificados por codones ricos en G y/o C, como Alanina, Glicina, Arginina y Prolina. Inversamente, los genes ubicados en L1 y L2 presentan una frecuencia más alta de aminoácidos codificados por codones ricos en A y/o T, como Fenilalanina, Isoleucina, Tirosina, Asparagina y Lisina. En otras palabras, esto explica lo que planteamos más arriba en el sentido de que los genes que están en L1 y H3, difieren mucho en el uso de codones y en los aminoácidos codificados.
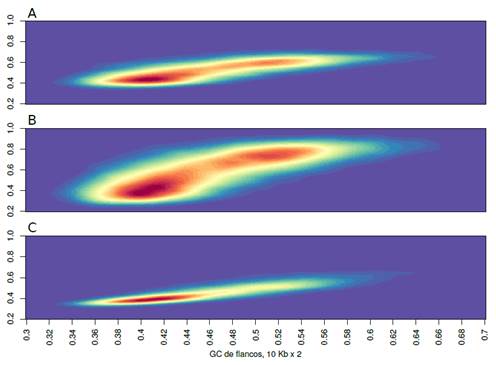
Fuente: elaboración propia.
Figura 6: se muestran los “hot-plots” de los contenidos de GC de exones (A), de GC3 (B) y de los intrones (C) (estos valores son mostrados en las ordenadas) en relación al GC de los isocoros donde se ubican los respectivos genes (abscisa). Las zonas más rojas son los sitios en el plano donde se ubican la mayor parte de los puntos, y las azules-celestes donde hay menos
Solamente como ejemplo, en la Figura 6 mostramos las correlaciones que existen entre GC de isocoros (eje x) y GC de exones (a), GC3 (b) e intrones (c) (eje y). Esta figura (plot de densidad) muestra en color rojo donde hay más puntos, y se va “yendo” hacia el azul donde hay menos. Algunas conclusiones que se pueden extraer son las siguientes. Primero, las correlaciones son siempre positivas y significativas, aunque, segundo, las pendientes cambian (ver más abajo). Esto último se debe a que, como ya hemos dicho, las fuerzas selectivas que operan sobre cada una de las variables mostradas en el eje “y” son diferentes, y nuevamente vemos que la que tiene mayor libertad es la posición 3 de los codones. Tercero, se aprecia claramente que los genes (Figura 6a) no se distribuyen igualmente a lo largo del genoma, y existen regiones más “cargadas” de secuencias génicas (los isocoros L y H2-H3).
Es importante tener en cuenta que este tipo de plot muestra en rojo solamente los sitios con más puntos en absoluto, sin embargo, para interpretarla debemos recordar que la cantidad de ADN es mucho menor en H2-H3 que en L, por lo tanto la cantidad de genes corregida por la frecuencia de cada isocoro, es mucho mayor, como ya habíamos visto más arriba, en las regiones ricas en GC. Al estudiar la Figura 6b se aprecia claramente la bimodalidad de la distribución de GC3 ya discutida más arriba.
Esta figura es complementaria de la Figura 5, pero agrega en qué regiones del genoma están ubicados los genes definidos por su GC3. Si comparamos las Figuras 6a y 6b, observamos que en la parte b (GC3) la pendiente es mayor es que en la a (GC de exones). Eso se debe a que los valores de GC3 pueden, por la libertad intrínseca a la estructura del código genético, aumentar (o disminuir) mucho más que el GC de los exones, en los que se incluye, naturalmente, el GC de las posiciones 1 y 2 de los codones, las que, como dijimos más arriba, son las más determinantes desde el punto de vista de los aminoácidos codificados.
Finalmente, en la Figura 6c se observa que la amplia mayoría de los intrones están ubicados en isocoros con un GC relativamente bajos, y a su vez no suelen alcanzar valores mayores a 50% de GC, lo cual se observaba en el histograma mostrado en la Figura 5b.
Resumiendo este punto, podemos concluir que a medida que los genes se ubican en isocoros más ricos en GC, todas sus partes (exones, intrones, las distintas posiciones de los codones tomadas de a una) van aumentando su GC; y lo hacen en forma diferente de acuerdo a la presión que sobre ellas ejerce la selección natural. Si bien no discutiremos a fondo este punto, es necesario aclarar que el sesgo mutacional del genoma humano (es decir, la tendencia hacia que un par de bases G:C mute hacia A:T y viceversa) es notoriamente hacia AT, lo cual explica, al menos en parte, por qué los intrones son notoriamente más pobres en GC que sus genes.
En este sentido, sucede lo mismo con los seudogenes.
Origen de los isocoros
Existen dos tipos de organización diferente en los vertebrados. Por un lado, los homeotermos (mamíferos y aves) presentan una heterogeneidad composicional marcada y tienen isocoros ricos en GC (siendo el genoma humano un ejemplo típico e igual al del resto de los mamíferos), mientras que los genomas de los poiquilotermos (peces, anfibios y reptiles) son menos heterogéneos y no presentan los isocoros H20. A su vez, estas características se reflejan en modelos diferentes cuando analizamos, en cada especie, los contenidos en GC de las posiciones sinónimas, exones, intrones, etc. (ver más arriba). Como es de esperar, las figuras correspondientes a los poiquilotermos muestran una dispersión menor de GC% y no llegan a valores tan altos como lo hacen los homeotermos. Por lo tanto, se puede afirmar que los patrones composicionales de aves y mamíferos son parecidos entre sí y, al mismo tiempo, diferentes del patrón poiquilotermo, tanto en los niveles de ADN como de secuencias codificantes. Dado que los mamíferos y las aves derivan de organismos de sangre fría (que se supone presentaban una organización en isocoros similar a la de los poiquilotermos actuales), se deduce que la mayor heterogeneidad composicional, y particularmente la aparición de los isocoros H, es coincidente con la aparición de los organismos de sangre caliente. Dicho con otras palabras, regiones definidas y discretas del genoma “poiquilotermo” se enriquecen en GC% en los genomas “homeotermos”. Por lo tanto, en la evolución de los genomas de los vertebrados ocurrieron dos “corrimientos” (transiciones) principales en los patrones composicionales: uno que tuvo como consecuencia el genoma tipo “mamífero” y el otro el genoma tipo “aves” (el cual es muy similar al patrón “mamífero” pero agrega el componente H4, es decir, presenta una familia de isocoros más rica en GC que los mamíferos). Es muy importante tener en cuenta que estas transiciones ocurrieron en forma independiente, ya que la evidencia paleontológica indica que los mamíferos derivaron de los terápsidos hace más de 200 millones de años, mientras que las aves aparecieron a partir de los dinosaurios unos 50 millones de años después. A las regiones del genoma de mamíferos y aves que todavía presentan el GC equivalente al de los isocoros de los organismos poiquilotermos (o sea, L1 y L2), se les llama “paleogenoma”, mientras que a las zonas que se enriquecieron en GC en los organismos homeotermos se les dio el nombre de “neogenoma”5.
El hecho de que son los mismos genes (y las mismas regiones genómicas) las que se enriquecieron en GC% en aves y mamíferos sugiere que las causas que determinaron estas transiciones pueden ser comunes. Se ha discutido mucho acerca de cuáles pueden ser estas causas; inclusive hay autores que postulan que el origen de los isocoros (particularmente los ricos en GC) no tiene ninguna causa selectiva. Revisaremos brevemente ambas posiciones, en primer lugar las esencialmente “neutralistas” (o sea que los cambios en GC no obedecen a ningún factor selectivo) y luego la “seleccionista”, la que, como su nombre indica, postula que el aumento en GC en determinadas regiones (H1, H2, H3 -y H4 en aves-) es el resultado de la acción de la selección natural.
A partir del descubrimiento de que distintos genomas bacterianos poseen diferentes composiciones nucleotídicas, se postuló21)(22) que las diferencias se debían a sesgos mutacionales en el sistema de replicación/reparación del ADN, o sea a diferencias en las tasas de mutaciones asociadas con cambios GC↔AT. Con distintas variaciones, Sueoka postula que esos sesgos mutacionales explican también la distinta composición nucleotídica intragenómica característica de los vertebrados, particularmente en aves y mamíferos. Entre las distintas objeciones que se han levantado contra esta hipótesis, creemos que hay dos muy fuertes. En primer lugar, los sesgos en los sistemas enzimáticos de replicación/reparación tendrían que haber ocurrido solamente dos veces en la evolución de los vertebrados, a saber, sólo en las líneas que dieron lugar a las aves y mamíferos, y jamás en todos los demás linajes que dieron lugar a los poiquilotermos contemporáneos. En segundo lugar, explicar de esta forma la aparición de los isocoros implica postular que dentro de los genomas de mamíferos y de aves existen no uno sino varios sesgos mutacionales diferentes operando en forma simultánea, por lo que se vuelve imprescindible postular desde esta óptica que distintas zonas del genoma son duplicadas/reparadas por distintas enzimas con distintos sesgos. Mencionemos, además, que el hecho de que los isocoros ricos en GC representen en aves y mamíferos la misma fracción del genoma (aproximadamente un tercio), a pesar de diferir el valor C (cantidad de ADN por genoma haploide) por un factor de tres, sería, desde esta óptica, una extraordinaria coincidencia.
Otra hipótesis muy aceptada actualmente es la que vincula la aparición de los isocoros H con la conversión génica23)(24)(25, fenómeno que se puede definir como el proceso por el cual una secuencia de ADN reemplaza a una secuencia homóloga de forma tal que las secuencias, luego del evento de conversión, son idénticas. La lógica de esta idea es que naturalmente el sistema enzimático de conversión génica tiende a cometer errores (con una frecuencia muy baja), los que están sesgados hacia G:C. O sea, si bien la tasa de errores que lleva desde un par A:T a uno G:C es muy baja, dado un tiempo evolutivo largo, las regiones del genoma en que más ocurra este fenómeno van a enriquecerse en GC. Y, como fue indicado más arriba, son las zonas más ricas en GC donde ocurren más eventos de recombinación (y de conversión génica). Si bien esta hipótesis parece muy razonable, al igual que la anterior no explica por qué la conversión génica “sesgada” ocurre solamente en mamíferos y aves, y no en los demás vertebrados. Esto resulta muy llamativo cuando el análisis de los genomas completos de peces, anfibios, reptiles, mamíferos y aves mostró que, en realidad, compartimos la aplastante mayoría de los genes...
La hipótesis seleccionista más conocida fue elaborada por el grupo de Bernardi26)(27)(5. La idea central es que las transiciones composicionales que llevaron a la aparición de los isocoros ricos en GC en mamíferos y aves se debe fundamentalmente a selección direccional, tanto positiva como negativa, actuando a nivel de los isocoros. A pesar de que las ventajas selectivas asociadas con los patrones composicionales pueden ser difíciles de identificar (no cabe duda que muchos factores deben estar actuando en forma simultánea), existe en la evolución de los vertebrados un hecho que podría explicar la aparición de los isocoros H. Efectivamente, el corrimiento composicional no ocurrió en alguno de los diversos pasos que caracterizaron la evolución de los vertebrados (de anamniotas a amniotas, de peces a tetrápodos, etc.) sino sólo y únicamente en las transiciones de poiquilotermos a homeotermos. Esto, afiman Bernardi y sus colegas, sugiere inmediatamente que uno de los factores principales para el cambio en los patrones composicionales fue el aumento de la temperatura corporal. El incremento en GC en los homeotermos parece lógico -en lo que a ventajas selectivas se refiere- ya que lleva a mayor estabilidad desde el punto de vista termodinámico, tanto en los niveles de ADN y ARN como de proteínas. Efectivamente, la riqueza en GC incrementa la estabilidad del ADN, ya que los pares de bases GC se unen por tres puentes de hidrógeno contra dos puentes de los pares AT; y esto ocurre no sólo en solución sino también a nivel de cromosomas, como lo indican las técnicas de bandeo R y T, que muestran que las regiones ricas en GC son más estables frente a la desnaturalización térmica que las bandas G, más pobres en GC. El referido aumento también tiene como consecuencia un incremento de la estabilidad térmica del ARN, ya que los transcriptos pueden adquirir una estructura secundaria más estable, y finalmente, a nivel de proteínas, los genes que están ubicados en zonas del ADN ricas en GC codifican niveles mayores de aminoácidos que confieren mayor estabilidad termodinámica (como arginina, alanina y glicina), y menos de los que la reducen (como serina y lisina).
A pesar de lo atractivo de esta hipótesis, es necesario remarcar que bajo ningún concepto postula que el aumento de la temperatura corporal sea el único factor que llevó a la aparición de los isocoros ricos en GC característicos de los homeotermos; simplemente pone el acento en una ventaja selectiva que resulta obvia, reconociendo, al mismo tiempo, que algo tan complejo como el fenotipo global del genoma debe ser necesariamente el resultado de múltiples factores que actúan en forma simultánea. Sin embargo, esta hipótesis tiene varios puntos en contra. Destacamos dos. En primer lugar, los homeotermos tienen temperaturas corporales que varían entre los 37º C y 42º C, y los poquilotermos suelen estar a alrededor de 15º C o 20º C, y la diferencia parece ser escasa desde el punto de biofísico como para explicar la aparición de los isocoros H. Por lo tanto, más allá de que no se discute la existencia de los isocoros, todavía no hay un consenso en cuanto a su origen y evolución.
Conclusiones generales: el genoma como un mosaico evolutivo
Para finalizar, nos parece importante señalar que los estudios sobre las propiedades composicionales del ADN de organismos multicelulares complejos, desarrollados fundamentalmente en los últimos 40 años, han mostrado en forma clara que el genoma es mucho más que la simple sumatoria de secuencias codificantes y no codificantes. Efectivamente, el genoma debe ser considerado como un sistema estructural, funcional y evolutivo integrado cuyas secuencias nucleotídicas están sometidas a reglas precisas que constituyen un “código genómico”. Esta teoría de la organización, fisiología y evolución del genoma asume que las propiedades composicionales de las moléculas de ADN (composición de bases, dinucleótidos y otras secuencias cortas) son características decisivas para la estructura, función y evolución del genoma.
En otras palabras, el genoma de los vertebrados no sería sólo un mosaico estructural y funcional (transcripción, duplicación, recombinación) de isocoros, sino que es, al mismo tiempo, un mosaico evolutivo, en el que cada región, definida por su composición de bases, se diferenciaría también de las otras por distintos niveles de restricciones y condicionantes evolutivas. Agreguemos un tema no menor: las secuencias “no codificantes”, que durante mucho tiempo fueron consideradas “secuencias egoístas”28)(29 o sea, secuencias cuya única “función” era perpetuarse a sí mismas en el genoma, hoy sabemos que son parte de un todo integrado y que evolucionan en equilibrio con el resto de las secuencias que constituyen el genoma.
Finalmente, queremos resaltar que nos encontramos en un momento crucial para comprender cómo se organiza, funciona y evoluciona el genoma. El desarrollo de técnicas nuevas de secuenciado, de aislamiento de transcriptos (ARNm) que se expresan en uno o muy pocos tejidos, y cuántos de ellos son efectivamente traducidos a proteínas y en qué cantidad, nos abren puertas imposibles de soñar sólo unos pocos años atrás. Este campo, tanto a nivel experimental como teórico está abriendo ventanas de oportunidades únicas para comprender cómo es nuestro material genético. Y por si fuera poco, el costo de secuenciar un genoma humano ha caído varios órdenes de magnitud, por lo que tener datos de miles de seres humanos individuales ya no es un posibilidad, sino una realidad. Todo apunta a que los próximos años serán sumamente excitantes en este campo, que afecta no solamente el conocimiento “básico” de nuestro genoma, sino que tendrá un impacto notable en la medicina.

