











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references

 Permalink
PermalinkIntroducción
La neurofibromatosis (NF) es el síndrome neurocutáneo más frecuente. Es una enfermedad hereditaria con afectación multisistémica1.
Se distinguen tres formas: NF tipo 1 (NF-1) o enfermedad de von Recklinghausen, la forma más común (85%); NF tipo 2 (NF-2) o forma central tipo adulto (10%), y NF tipo 3 (NF-3) o forma mixta, excepcional1.
La prevalencia estimada de NF-1 es 1 en 3000 recién nacidos. Se hereda con carácter autosómico dominante con penetrancia completa y expresividad variable. El gen responsable está localizado en el cromosoma 17q11.2 y codifica la neurofibromina que tiene un papel modulador en el crecimiento celular y la diferenciación de la cresta neural. Actúa como supresor tumoral. El 50% de los casos corresponden a mutaciones de "novo"1.
Las manifestaciones cutáneas están presentes en casi todos los pacientes. Son típicas las máculas hiperpigmentadas “manchas café con leche” en número y tamaño variables que generalmente se detectan en los primeros meses de vida. Otras lesiones cutáneas frecuentes son las manchas lentiginosas (freckling) en axilas, ingles o cuello, de aparición más tardía (entre 3 y 5 años)1.
Los neurofibromas y los gliomas ópticos son los tumores más frecuentes. Los neurofibromas pueden ser plexiformes o cutáneos. Los plexiformes están presentes en el 25% de los individuos con NF1 y son típicamente congénitos1)(3)(4)(5)(6.
La incidencia de neoplasias en los pacientes con NF-1 es de un 7%7.
El diagnóstico de NF-1 se basa en criterios clínicos que son muy específicos en niños mayores o adultos (97% cumplen con los criterios a los 8 años de edad). En niños más pequeños el diagnóstico es más dificultoso, ya que al año de edad el 46% de los pacientes no los cumplen. En ellos, el diagnóstico definitivo se puede demorar hasta los 4-5 años, cuando la enfermedad ya adquiere mayor expresividad1)(8)(9. En la Tabla 1 se muestran los criterios diagnósticos de NF1 elaborados por el Instituto Nacional de la Salud de EE.UU (1987). Es necesario para el diagnóstico la presencia de dos o más de los mismos10.
Tabla 1: Criterios diagnósticos de neurofibromatosis tipo 1Criterios Diagnósticos de NF-1
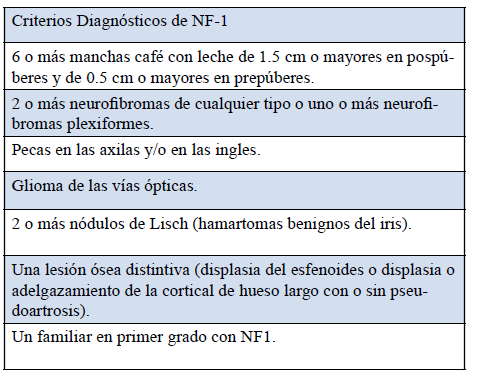
Fuente: Instituto Nacional de la Salud de EE.UU (1987)
En la actualidad se dispone de estudios genéticos para el diagnóstico de NF1. Son utilizados para confirmar el diagnóstico en pacientes que cumplen sólo con un único criterio diagnóstico; cuando la presentación clínica es inusual; para realizar screening prenatal y consejería genética11.
No se dispone de tratamiento para esta enfermedad. El seguimiento clínico es fundamental para diagnosticar y tratar oportunamente las complicaciones1)(2.
El objetivo de esta comunicación es describir una niña de 3 años con NF- 1 con una complicación tumoral de baja frecuencia que representa un desafío terapéutico debido a la topografía y frecuencia de recidiva. En el caso clínico analizado el tumor recidivó, por lo que fue necesaria una reintervención del mismo.
Caso Clínico
Tres años, sexo femenino, sin antecedentes familiares ni perinatales a destacar. Antecedentes personales: NF 1 de reciente diagnóstico con manifestaciones cutáneas, sin afectación ósea, ocular ni neurológica previa.
Ingresa por hemiparesia derecha. Comienza dos meses previos con dolor a nivel de miembro superior derecho irradiado a falanges distales, disminución de fuerzas, y limitación funcional progresiva. En la evolución agrega alteración de la marcha, con disminución de la movilización de miembro inferior derecho, sin dolor No antecedentes de traumatismos previos, no fiebre ni otra sintomatología.
Al examen buen aspecto general. Piel: diez manchas café con leche en tórax, abdomen, región lumbar y miembros inferiores, con diámetros entre 1 y 4 cm. Pecas en axilas. Mucosas sin alteraciones. Del examen neurológico se destaca: Sector espinal: Preferencia en el uso del miembro superior izquierdo. Miembro superior derecho: paresia distal, hiperpasividad, dolor a la movilización, reflejo bicipital disminuido. Miembro inferior derecho: Paresia que vence gravedad, hipopasividad, reflejo rotuliano vivo con aumento del área. Babinski. Marcha: parética, en hoz. Resto del examen sin alteraciones.
Se solicita Resonancia Nuclear Magnética (RNM) de columna cervical, dorsal y lumbar que evidencia lesión sólida, intrarraquídea, localizada en raquis cérvico-dorsal, desde C6 a T1, que compromete el espacio intradural a derecha, extendiéndose al foramen C6-C7, el cual está aumentado de calibre. En este nivel el proceso adopta el aspecto en reloj de arena, comprimiendo el eje medular, el cual se encuentra lateralizado a izquierda, de señal conservada. La lesión es bien limitada con una extensión total de 22cm. Se presenta isointensa con el tejido muscular en las secuencias T1 e hiperintensa en las secuencias T2. Luego de la administración del contraste intravenoso se observa intenso realce del mismo. Los segmentos vertebrales y los discos intervertebrales cervicales, dorsales y lumbares son de aspecto y señal conservadas. El cono medular se localiza a la altura de L1. En suma: lesión sólida intradural, extramedular, centrada en C6-C7, con extensión al foramen derecho.
Se realiza neurocirugía con exéresis tumoral. Se identifican nódulos tumorales en toda la extensión del músculo paraverterbral. Se reseca en forma subtotal el componente extrarraquídeo, quedando pequeño remanente adherido a la arteria vertebral.
Anatomía patológica: proliferación mesenquimática fusocelular, en tejido muscular con nódulos bien limitados interpuestos en haces musculares, y en tejido intradural con células serpenteantes y núcleos fusiformes compatible con neurofibroma. Inmunohistoquímica: proteína S100 positiva.
En el postoperatorio inmediato, buena evolución clínica, con recuperación progresiva de la hemiparesia. Se otorga alta con gabapentina y dexametasona vía oral en descenso y control programado en neurocirugía, fisiatría y oncología.
Al año, presenta recidiva de neurofibroma intrarraquídeo a nivel de C5-C6 con compresión medular a nivel de C6-T1, por lo que se realiza nueva exéresis tumoral.
Discusión
La paciente que se comunica presenta un tumor de baja frecuencia en la infancia. El mismo es de localización intradural, extramedular. Los tumores con esta topografía representan el 15% de los tumores raquimedulares, los neurofibromas y neurinomas constituyen dos tercios de los casos1)(5.
Los neurofibromas frecuentemente se diagnostican en pacientes con neurofibromatosis, si bien existen descripciones de tumores solitarios en pacientes sin la enfermedad. La paciente analizada presenta un neurofibroma de raíz espinal. Los mismos pueden surgir en la médula y en múltiples niveles, y probablemente representan tumores plexiformes dada la participación de múltiples fascículos nerviosos. Los neurofibromas plexiformes característicamente involucran una cantidad significativa de un único nervio, o más comúnmente, infiltran totalmente un plexo nervioso, invadiendo estructuras vasculares y viscerales. Esto impide la resección completa del tumor, tal como sucedió en este caso. El abordaje de estas lesiones es complejo además porque presentan un comportamiento biológico impredecible, ya que algunas lesiones exhiben crecimiento y sintomatología progresivos, mientras que otras permanecen estables y relativamente asintomáticas por largos períodos de tiempo2)(5)(12.
En el seguimiento de los niños con NF-1 la indicación de estudios de imagen es controversial, dado que la mayoría de las lesiones no van a ser pasibles de tratamiento y son de crecimiento lento. Las guías de consenso actuales no recomiendan imágenes de rutina. Se indica el control clínico e imagenológico de las lesiones cuando estas comienzan a producir síntomas tal como sucedió en este caso2)(13)(14.
Debido a las características infiltrativas y la diseminación paraespinal extensa de los neurofibromas plexiformes, los pacientes presentan síntomas de mielopatía o radiculopatía multisegmentaria secundario al compromiso de múltiples niveles espinales adyacentes. La resección de estas lesiones generalmente presenta morbilidad asociada al compromiso de estructuras críticas extraespinales15.
En el interior de los neurofibromas plexiformes pueden gestarse tumores malignos de la vaina nerviosa, muchas veces no detectados hasta la aparición de las manifestaciones de sus metástasis. Por este motivo se recomienda la realización precoz de biopsia o resección en lesiones de rápido crecimiento o frente a la presencia de cambios clínicos y/o dolor16.
Existen pocos reportes de casos e información limitada en la literatura sobre presentación clínica, manejo y pronóstico de este tipo de pacientes.
En 2015, Safaee y cols. en un estudio retrospectivo analizaron la relación entre localización de los tumores de la vaina de los nervios espinales, la extensión de la resección, el diagnóstico de neurofibromatosis, y el resultado clínico. En esta serie, los neurofibromas representaron el 24% de los tumores siendo su topografía más común la médula cervical (74%). La tasa de resección total fue baja y la recurrencia o progresión elevada (17%)17.
En un estudio multicéntrico realizado por Leonard y cols. entre 1996 y 2006, se identificaron trece pacientes con compresión de la médula cervical debida a neurofibromas plexiformes, con edades entre 9 y 61 años; siendo cuatro pacientes menores de dieciocho años. El déficit neurológico progresivo motivo la cirugía en 11 pacientes. Se observó que el riesgo de presentar síntomas debido a un neurofibroma no decrece con la edad. La presentación más común fue: cuadriparesia progresiva, paraparesia, incontinencia y dolor cervical. Los segmentos involucrados más frecuentemente fueron C2 y C3. Excepto un paciente, el resto presentaron secuelas con déficits motores o sensitivos menores6.
En nuestro medio, no se dispone de comunicaciones de casos con neurofibromas plexiformes intradurales. Figura 1.
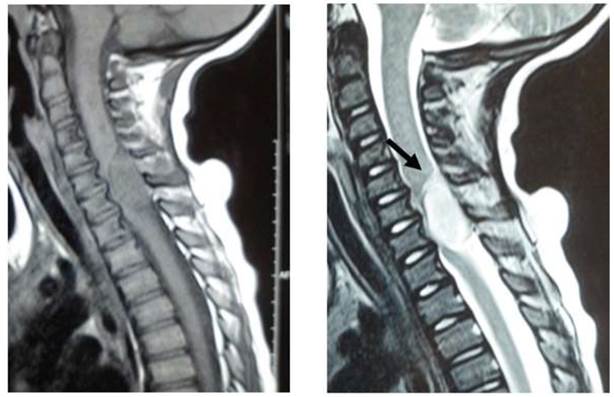
Conclusión
La NF1 es un trastorno de herencia autosómica dominante, progresivo y multisistémico. Es una enfermedad compleja que requiere un control evolutivo fundamentalmente clínico, y de ser necesario imagenológico para detectar y abordar oportunamente los tumores, su complicación más importante. Los neurofibromas plexiformes representan el 25% de los tumores que desarrollan estos niños. La localización intradural, extramedular es inhabitual en la infancia y representa un gran desafío terapéutico debido a la elevada morbilidad asociada y las frecuentes recidivas.

