










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkOdontoestomatología
versión On-line ISSN 1688-9339
Odontoestomatología vol.12 supl.16 Montevideo dic. 2010
SINDROME KID. ABORDAJE INTERDISCIPLINARIO.
Dra. Carla De Negri *
*Asistente, Grado 2, Programa Docencia-Servicio, Área del Niño. Colaborador Honorario. Cátedra de Odontopediatría. Facultad de Odontología. UDELAR.
Resumen
Se presenta el caso clínico, de una niña de 2 años de edad, nacida en el Centro Hospitalario Pereira Rossell, portadora del Síndrome KID. Se trata de una displasia congénita ectodérmica caracterizada por la asociación de queratitis, ictiosis y sordera. Es producida por un desorden autosómico dominante, esporádico. Algunos autores lo refieren asociado a la presencia de alteraciones estructurales de órganos dentarios, del tipo hipoplasia e hipomineralización del esmalte y también a gingivitis y candidiasis a nivel de la mucosa bucal. La intervención oportuna del odontopediatra, integrando el equipo de salud, brinda un aporte importante, en la búsqueda de estrategias terapéuticas adecuadas al paciente y al contexto asistencial. El abordaje de tratamiento en pacientes portadores del Síndrome KID debe ser multidisciplinario, requiriendo en niños, la integración de un equipo de salud con pediatra, dermatólogo, oftalmólogo, otorrinolaringólogo y odontopediatra.
Palabras claves
Queratitis, ictiosis, sordera, Síndrome KID.
Kid Syndrome. An interdisciplinary approach.
Abstract
The clinic case of a two year old girl, born in the Pereira Rossel Hospital, with KID Syndrome is presented. It is a congenital, ectodermic dysplasia, characterized by the association of keratitis, ichtyiosis and deafness. It is produced by an autosomic dominant disorder. It can be associated to the presence of structural alterations in tooth: enamel hipoplasia and hipomineralization, as well as gingivitis and oral candidiasis in oral mucous membrane. The opportune intervention of the pediatric dentist gives an important help in the search of the adequate therapeutic strategies for the patient and the assistential context. The treatment for people with KID Syndrome has to be multidisciplinary, requiring in children a health team formed by pediatric physician, dermatologist, ophthalmologist, otorhinolaryngologist, and pediatric dentist.
Key words
Keratitis, ichtyiosis, deafness, KID Syndrome.
Fecha de recibido: 02.09.10
Fecha de aceptado: 11.11.10
Introducción
En el marco de la Carrera de Especialista en Odontopediatría, de la Facultad de Odontología de la Universidad de la República, Módulo 3, que se implementa en el Centro Hospitalario Pereira Rossell, cursantes y docentes se integran a un equipo de salud estable para el abordaje terapéutico de los pacientes allí internados.
El Sindrome KID fue descripto por primera vez en 1915 por F.S. Burns (1-5) aunque su designación no es conocida hasta 1981 con la publicación de B.A. Skinner y col en Archivos de Dermatología.
KID se trata de un acrónimo en inglés de: keratitis (K), ictiosis (I), deafness(D). Consiste en una displasia ectodérmica congénita, producida por un desorden autosómico dominante esporádico que afecta a la epidermis, el epitelio corneal y el oído interno Se caracteriza por la asociación de queratitis, ictiosis (piel extremadamente seca) y sordera. Las lesiones dérmicas son hiperqueratósicas, presenta alopecia cicatrizal, distrofia ungueal, anomalías dentarias y neovascularización corneal progresiva (1-3, 5-12).
El objetivo de este trabajo, es sensibilizar al profesional de la salud sobre la necesidad del abordaje multidisciplinario en pacientes portadores de patologías crónicas de alta complejidad, tomando como punto de partida el estudio de un caso clínico de una niña de 2 años portadora del Síndrome KID. Si bien las características a nivel general son de relevancia, hasta con riesgo de vida, la temprana y oportuna intervención del odontopediatra, brindará un tratamiento preventivo-terapéutico para las patologías bucales que acompañan al síndrome, colaborando en mejorar la calidad de vida y el confort del paciente.
Se realizó una revisión bibliográfica utilizando como fuentes: MEDLINE, PMID, LILACS ID e IBECS ID, en el período 1987-2010, encontrándose reportados unos 100 casos en toda la bibliografía médica y solamente dos artículos en Latinoamérica.
Antecedentes
La presentación del Síndrome KID es esporádica y presenta distribución similar en ambos sexos. Los cambios en la piel se manifiestan al nacimiento o durante la infancia, siendo lo más frecuente en los tres primeros meses de vida, con períodos de exacerbaciones y remisiones (5). Se presentan como lesiones eritematosas costrosas en el cuero cabelludo, tronco y zonas de flexión, algunas veces eritematosas e hiperqueratósicas. Pueden observarse placas verrugosas en: frente, mejilla, zona perioral, codo, rodilla y pericráneo. Otros rasgos cutáneos que pueden presentar son: hiperqueratosis palmoplantar (41%), uñas distróficas y alopecia parcial o total (79%) (1, 2, 7, 9,11). También se han reportado carcinomas de piel, así como otros tumores malignos. (2, 4, 5, 9, 11).
El conocimiento de este síndrome, permite tempranamente evaluar problemas oftalmológicos y auditivos. Las lesiones oculares del Síndrome KID se expresan más tardíamente que el resto de las alteraciones y aunque suelen detectarse en la infancia, pueden no cursar con sintomatología hasta la pubertad (3). Las alteraciones oculares suelen ser conjuntivitis, fotofobia y/o abrasiones de córnea, pudiendo en algunos casos llegar a la ceguera (1, 2, 11). En referencia a la sordera neurosensorial es congénita y no progresiva, pudiendo presentarse al nacimiento, como también en los dos primeros meses de vida. En una revisión de 61casos, Cáceres Ríos et al. (1993), constataron sordera, en el 100% de los pacientes (1,5). Las manifestaciones neuromusculares son poco frecuentes, presentándose en algunos casos, retraso sicomotriz. La mayoría de los pacientes son intelectualmente normales, sin embargo, se han descripto casos asociados a hipoplasia cerebelar (2, 4, 13).
Los portadores del Síndrome KID cursan frecuentemente infecciones bacterianas, virósicas y fúngicas tanto cutáneas como de mucosas, siendo la Candidiasis la infección oportunista más frecuente, reportándose en el 50% de los casos, según algunos expertos asociado a la inmunodeficiencia intrínseca (1, 2, 4, 5, 9, 11). También se informa sobre la presencia de gingivitis de fácil sangrado (1, 3-5). Algunos autores han reportado la presencia de leucoplasia en la mucosa bucal, carcinoma de células escamosas y fisuras profundas en la lengua, pero aclaran el carácter incierto de que estas lesiones sean características del síndrome (5). La literatura revisada describe la presencia de patologías dentarias asociadas a la alteración epitelial como ser hipoplasia e hipomineralización adamantina. Estos defectos aumentan el riesgo a desarrollar lesiones de caries así como al desgaste de las estructuras dentarias y la hipersensibilidad. Dado que el Síndrome KID compromete tejidos de origen ectodérmico como epidermis, epitelio corneal, oído interno, dientes y uñas, ha sido considerado como un defecto ectodérmico congénito más que una verdadera ictiosis (1). Las ictiosis son un grupo de enfermedades que producen escamas visibles en toda o gran parte de la superficie de la piel, de ahí su nombre que significa pez.
Desde el punto de vista etiopatogénico, se ha informado la transmisión madre-hija, lo que sugiere un carácter autonómico dominante (2-5). El Síndrome KID es causado por mutación espontánea del gen GJB2, que codifica la conexina 26 (CX26), así como también mutación en el gen GJB6, que codifica la conexina 30 (CX30) (2-4, 11-15). La conexina 26 es una proteína esencial para las formaciones de unión tipo gap junction de las células epiteliales. Una gap junction es una firme cadena con muchos canales intecelulares que controlan y coordinan gran variedad de señales celulares, permitiendo el paso de pequeñas moléculas como iones, metabolitos y otras moléculas señalizadoras entre células. Las mutaciones erróneas CX26, han demostrado causar queratitis, ictiosis y sordera (KID), síndrome palmoplantar, queratoderma, asociado con pérdida de audición. Las mutaciones en el gen de la conexina 26 (CX26), locus DFMB1, en el brazo largo del cromosoma 13, dan cuenta del 60% de hipoacusias con herencia recesiva, de 20% de las sorderas infantiles y se han encontrado en portadores sanos de poblaciones caucásicas en una frecuencia que oscila entre 1 y 2,8%. Aunque la distribución de la conexina 26 en la córnea no ha sido aún investigada, el daño de las gap junction dependiente de la red epitelial corneal es posible que sea el responsable del trastorno de la superficie ocular (3). La presencia de una mutación patogénica del cromosoma CX26 y CX30, da características semejantes al Síndrome KID por lo que sugiere la heterogeneidad genética de la patología pudiendo causar diferentes fenotipos.
Descripción del caso clínico

MS, 2 años de edad, sexo femenino
Fecha de nacimiento: 27.04.2007
Talla: 34 cm. Peso: 3.415 mg.
Nacimiento: Quinta gesta, por parto vaginal, de 38 semanas, permaneciendo 3 meses en incubadora. Diagnóstico al nacimiento: Síndrome KID, Ictiosis congénita, Cardiopatía congénita e insuficiencia mitral, anacusia y ceguera total.
Madre: CM, 27 años de edad.
Hermanos: 4, 7, 10 y 11 años, sanos.
La paciente ingresó al Centro Hospitalario Pereira Rossell, el 24 de abril 2009 (Fig. 1).
Motivo de consulta: rinorrea mucosa y tos acuosa de 48 hs. de evolución. Hipertermia (40º). Secreciones seropurulentas en lesiones de piel, de región de glúteo, cuello y axila.
Se diagnosticó desnutrición crónica, anemia, conjuntivitis, queilitis, candidiasis en mucosas y piel.
Al examen clínico presentó: ictiosis de piel, placas eritematosas e hiperqueratosicas, cándidiasis mucocutánea crónica, que compromete cuero cabelludo (alopecia), zonas de flexión, palmoplantar; distrofia de uñas en manos y pies, abdomen abultado, globuloso. Asimismo se informó retraso general del crecimiento y comportamiento correspondiente a 6 meses de edad. (Figuras 2-5)
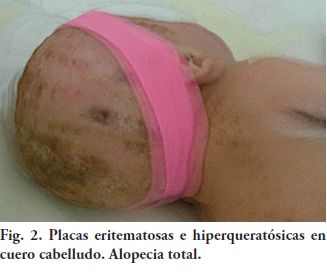
Fig. 2. Placas eritematosas e hiperqueratósicas en cuero cabelludo. Alopecia total.
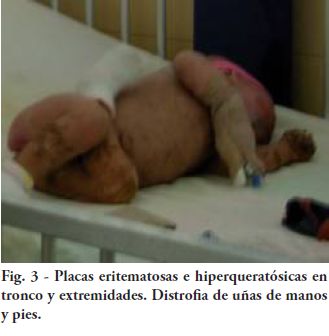
Fig. 3 - Placas eritematosas e hiperqueratósicas en tronco y extremidades.
Distrofia de uñas de manos y pies
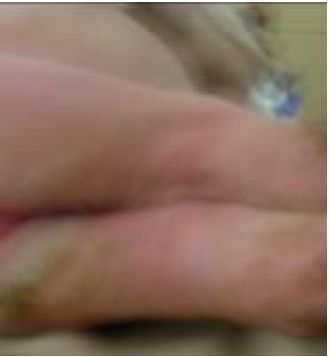
Fig. 4 - Secreciones seropurulentas en lesiones de piel en miembros inferiores
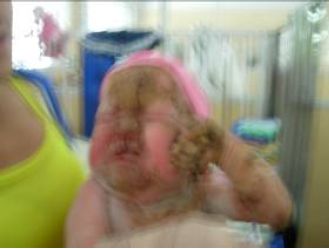
Fig. 5 - Placas verrugosas hiperqueratósicas en frente, nariz, mejillas
labios y mentón. Uñas distróficas en manos.
Al examen bucal se observó: 20 dientes temporarios con alteraciones de estructura del esmalte (de acuerdo a la clasificación de la FDI de 1982, hipoplasia Tipo 6 (defecto hipoplásico en que el esmalte está totalmente ausente), con gran abrasión, exposición dentinaria en todas las piezas dentarias e hipersensibilidad, presencia de gingivitis generalizada de muy fácil sangrado y candidiasis seudomembranosa en paladar, dorso de lengua y piso de boca.
En el interrogatorio la madre relató que los incisivos temporarios erupcionaron a los seis meses de edad, que presentaron color blanco y eran largos pero que en poco tiempo se desgastaron y tomaron un color amarillento. Al intentar introducirle los alimentos sólidos con cuchara, manifestaba dolor en los dientes. Sus encías siempre eran sangrantes. La niña se alimentó sólo con mamadera. La madre licuó toda la alimentación, balanceada. No tenía buena prensión oral de la mamadera, hecho que provocaba la ingesta de mucho aire y de poco alimento. No permitió ayuda en la colocación del biberón, ni que se la tomara en brazos para ser alimentada.
Se realizó diariamente en su domicilio, indicado por el equipo médico, el siguiente tratamiento:
-
en la piel - aplicación de crema dermatológica compuesta por Agua Termal 60% (Roche-Posay), cera 10% (Karité) y glicerina 7% (Lipikar), una vez al día, para disminuir la irritación y lubricarla. Control dermatológico cada 15 días.
-
en la boca: nistatina gel, cada 6 hs. previo lavado con una gasa y agua hervida y secado. La colocación del gel se realizó en paladar, dorso de lengua y piso de boca; para el tratamiento de la candidiasis.
Durante el período de internación el tratamiento médico consistió en la administración de Clindamicina intravenosa por tres días, control del peso e hidratación. Se realizaron curaciones en las zonas afectadas y nistatina, 0,5 cm3 cada 6 hs. por siete días para la candidiasis bucal.
Desde el punto de vista del tratamiento odontopediátrico se planteó la realización de un tratamiento sintomático con el objetivo de mejorar la calidad de vida de la paciente, permitiendo que se alimentara mejor y de educación para ayudar a la familia en los cuidados domiciliarios. Se realizaron aplicaciones profesionales de caseín-fosfopétido (CPP) con fosfato de calcio amorfo (ACFP), (Recaldent™) y se indicó su utilización a domicilio con el objetivo de favorecer la remineralización y disminuir la sensibilidad dentaria. También se aplicó digluconato de clorhexidina, en forma de gel, con el fin de controlar la flora bucal (Streptococcus Mutans y Candida Albicans) y la inflamación gingival.
Discusión
El Síndrome KID, es un trastorno poco frecuente, del cual se han publicado apenas un centenar de casos, en la literatura médica.
La mayoría son esporádicos y se distribuyen de igual forma en ambos sexos, afectándolos de igual manera. Las edades reportadas son desde el nacimiento, hasta 33 años de edad. Las características clínicas son, queratitis, ictiosis y sordera neurosensorial, presentándose frecuentemente una variedad de anomalías asociadas.
Es importante destacar que nuestro paciente presenta desde su nacimiento, todas las características clínicas propias del síndrome, agravada por una cardiopatía congénita del tipo CIV-CIA (comunicación interventricular e interauriocular), retraso general del desarrollo y desnutrición severa. En la literatura revisada se cita la presencia de alteraciones estructuras de las piezas dentarias, del tipo de hipomineralizaciones e hipoplasias del esmalte y caries activas; la presencia de candidiasis crónicas y gingivitis (5, 9). El paciente descripto, presenta 20 piezas temporarias con hipoplasias e hipocalcificaciones, con abrasión y exposición dentinaria en todas las piezas dentarias e hipersensibilidad. Gingivitis de fácil sangrado y placas de Cándida Albicans en paladar, dorso de lengua y piso de boca, en forma crónica. La hipoplasia del esmalte se define como uno de los defectos del desarrollo de los tejidos duros del diente que ocurren antes de la erupción del mismo como el resultado de un trastorno en la formación del esmalte (durante el proceso de amelogénesis). La etapa de calcificación de la dentición temporal comienza entre los cuatro y seis meses de vida prenatal y termina en la zona coronaria al año de edad, así cualquier trastorno de la matriz del esmalte origina un defecto hipoplásico. Estos defectos o anomalías varían en gravedad y se manifiestan clínicamente en su forma mas leve como pequeñas manchas blancuzcas opacas aisladas y diminutas fositas hasta manchas marrones y fosas y escotaduras marcadas que dan al diente un aspecto de corroído. Los posibles factores causales de esta alteración de la amelogénesis son muy numerosos, distinguiéndose tres tipos de agentes: las anomalías hereditarias, los traumas localizados y los factores sistémicos (16). El diagnóstico temprano es fundamental. De acuerdo a los casos estudiados el pronóstico general es bueno y el seguimiento de por vida de estos pacientes es necesario, porque puede estas asociado con tumores malignos especialmente carcinoma de células escamosas de la piel. Sin embargo el paciente por las características antes descriptas, padece un sufrimiento significativo y una calidad de vida muy deteriorada, con compromiso de vida. Es así, que la intervención del odontopediatra, podría haber aportado tempranamente, tratamientos alternativos y preventivos, con la posibilidad de enlentecer o detener el deterioro del estado de su salud bucal. Los tratamientos reportados en la bibliografía van dirigidos exclusivamente a solucionar las alteraciones de piel, ocular y visual. En los 100 artículos revisados no se reportaron casos donde se establezca la intervención del odontólogo. Si bien el abordaje es multidisciplinario, estaría indicado incorporar al odontopediatra, a los efectos de aportar soluciones a las patologías prevalentes en boca y mejorar la calidad de vida de estos pacientes.
Para nuestra paciente se plantió la realización de un tratamiento sintomático, con el objetivo de aliviar y reducir los síntomas, permitiendo una mejor alimentación y de educación y apoyo a la familia, con énfasis especial en la atención en domicilio. Se realizan aplicaciones profesionales de caseín-fosfopétido (CPP) con fosfato de calcio amorfo (ACFP) (Recaldent™) en crema y se indica su utilización a domicilio con el objetivo de favorecer la remineralización y disminuir la sensibilidad dentaria. También se indica la aplicación de Digluoconato de Clorhexidina en forma de gel, con el fin de controlar la flora bucal (Streptococcus Mutans y Candida Albicans) y la inflamación gingival.
CPP-ACFP- Representa un nanocomplejo derivado de la proteína de la leche, caseín fosfopéptido (CPP) con fosfato de calcio amorfo (ACFP) con una concentración al 10% más 900 ppm de Fluor (MI Paste Plus™ con Recaldent™). Se adhiere a los tejidos blandos, la placa, la película y la hidroxiapatita, proporcionando los minerales incluidos en su composición en forma amorfa, tanto a la saliva como al líquido extracelular de la placa microbiana. La aplicación de este compuesto, además de incrementar los niveles de calcio y fósforo biodisponibles, amortigua los cambios de pH en placa y disminuye la adherencia y el crecimiento de Sstreptococcus Mutans y Sobrinus. Es relevante entonces su acción remineralizante, liberando lentamente sus componentes a nivel de superficie dentaria y difundiendo a nivel sub superficial (17).
DIGLUCONATOO DE CLORHEXIDINA- Es el agente antimicrobiano bucal más usado, efectivo y documentado en eliminar o disminuir la flora estreptocócica en procesos dependientes de estos microorganismos. La eficacia antimicrobiana de la clorhexidina permite controlar otras bacterias orales siendo efectivo en el control de hongos como la Candida Albicans (18-19). La aplicación de digluconato de clorhexidina en una concentración del 0,12% y en forma de gel (Bucogel ®), es de muy fácil aplicación por el profesional y por la familia del paciente. La dosis tóxica presenta un alto margen con la terapéutica, siendo de esta manera de elección para el tratamiento de un niño pequeño (20).
Conclusión
Por todo lo expuesto se concluye que el abordaje para la atención de los pacientes con Síndrome KID debe ser multidisciplinario, en cuanto a planificación como a la aplicación de las medidas terapéuticas. Las consultas regulares al dermatólogo son necesarias por la relevancia de las lesiones cutáneas y la posible presencia de carcinoma de células escamosas, aunque poco frecuentes. Asimismo, acuden regularmente al oftalmólogo por sus conjuntivitis recurrentes y al otorrinolaringólogo. Hemos evidenciado la importancia de la integración del odontopediatra al equipo de salud, mejorando la calidad de vida del paciente, disminuyendo los síntomas y brindando a su familia la posibilidad de realizar tratamientos sencillos y efectivos. La incorporación del odontólogo al equipo multidisciplinario y el establecimiento de estrategias de tratamiento específico, adecuado a las características del paciente, de su medio familiar y del contexto de atención colaborarán en el mejoramiento de la calidad de vida de los portadores de síndromes complejos como el desarrollado en el presente trabajo.
Referencias bibliográficas
-
Caceres Rios H., Baiona R., Castro R. I., Velásquez F. ¿Que síndrome es? Servicio de Dermatología del Instituto Especializado de Salud del Niño, Lima-Peru. Dermatol Pediatr Lat 2004; 2(2): 156-159.
-
Susan B. Mallory, MD y Bernice R. Krafchik, MB, Ch.B., FRCPC. What Síndrome is This? Departamentos de Dermatología y genética Médica, el Hospital for Sick Children, Toronto, Canadá. Dermatology Pediatric 2006;23 (1): 81-83.
-
Gómez Faiña P; Ruiz Viñals AT; Buil Calvo JA; España Alvelda a; Pazos Lopez M; Castilla céspedes M. Patient with severe corneal disease in kid síndrome. Arch soc esp oftalmol 2006; 81: 225-228.
-
Gonzalez ME, Tlougan BE, Price HN, Patel R, Kamino H, Scraffer JV.Department of Dermatology, New Cork University, USA. Keratitis-ichthyosis-deafness (KID) síndrome. Dermatol Online Journal . 2009;15(8):11-13.
-
L Miteva, MD, PhD. Keratitis, Ictiosis and Deafness (KID) Síndrome. Dem.Pediatr. (Department of Dermatology and Venerology Medical University, Sofia, Bulgaria. 2002; 19(6); 513-515.
-
Alonso,A.B de; Cesarios, G; Pecoraro, V; Proske,A. Síndrome Kid (Queratitis-Ictiosis-sordera) con porosas ecrinos multiples. Rev. Argent. Dermatol. 1993; 74(1):10-4.
-
Caceres Rios, H. M.D.; Tamayo Sanchez, L. M.D..; Duran Mckinster, C. M.D,Orozco, M.M.D.; Ruiz M., Ramon M.D. Keratitis,Ictiosis, and deaffness (KID síndrome): Review of de literatura and proposal of a new terminology. Pediatric Dermatology. 1996; 13(2):105-113.
-
Burgos, G.; Cohen Sabban, E.; Tous, V.; Sethman, A.; Casas, G.; Cremona, G.; Allevato, M.; Cabrera, H.. Síndrome KID: presentación de un caso y remisión de la literatura. Arch argent. Dermatol. 2005;55(3):109-113.
-
Ali Abdollahi MD,Masoumeh Barzegari MD, MaryamAkhyani MD, Izabas Toosi Md, Alireza Miresmaili MD. KID Síndrome. Dermatology Online Journal. 2007; 13(4): 11.
-
Harunak, Suga Y; Oizum A, Mizumo Y, Endo H, Shimizut, Hasegawa T, Ikeda S. Severe form of Keratitis-ichthyosis-defness (KID) síndrome associated with septic complications. J. Dermatol. 2010; 37(7): 680-2.
-
Amy S. Paller and Anthony j. Manzini. Hereditary Disorders of the dermis. En: Hurwitz. Clinical Pediatric Dermatology. A textbook of skin disorders of childhood and adolescente. 3ª edition, China. Elsevier Sauders, 2006. cap. 5, p.115-116.
-
Josef G. Morelli. The skin desorders of keratinization. En: Nelson. Textbook of Pediatric. Kliegman, Behuman, Jonson, Stanton. 18º edition, China, Sounders Elsevier, 2007. Cap.657, p.2709.
-
Todt I, Mazereeuw-Hauter J, Binder B, Willems PJ. Department of Otolaryngology, Unfallkrankenhaus Berlin, Charite Medical School, Berlin, Germany. Dandy-Walker malformation in patient with KID síndrome associated whith a hetetozygote mutation in the GJB2 gene encoding connexin 26. Clin Genet 2009. 76(4): 404-8.
-
Lazic T, Horii KA, Richard G, Wasserman DI, Antaya RJ. Department of Internal Medicine, Yale University School of Medicine, New Haven, Connecticut, USA. A report of GJB2 connexin 26 mutation in two patient- a new subtype of KID síndrome. Pediatr Dermatol 2008; 25(5): 535-40.
-
Sbidian E, Feldmann D, Bengoa J, Fraitag S, Abadie V, de Prosa Y, Bodemer C, Hadj-Rabia S. Department of Dermatology, MEGEC, Necker-Enfants Malades Hospital, AP-HP, Paris, Francia. Germline mosaicism in Keratitis- ictiosis-desfness síndrome: pre-natal diagnosis in a familial letal form. Clin Genet. 2010; 77(6): 587-92.
-
Trancho G.J., Robledo B. Patología Oral: Hipoplasia del esmalte dentario. Departamento de Biología AnimalI (Antropología) Facultad de Biología. Universidad Complutensede Madridna.
-
López MC, Castro J. La terapia remineralizadora en la práctica preventiva y restauradora de la odontología. Odontoestomatología Dic.2008; vol .X, nº 11; 22-30.
-
Ismael Yévenes L, Juan Reyes Y, Nestor Campos P, Victor Saragoni F. Efecto inhibitorio en la placa microbiana y propiedades antibacterianas de enjuagatorios de clorhexidina. Av Periodon Implant. 2003 ; 15(1):19-24.
-
Matos BM, Deco CP, Olivera LD, Jorge AOC,Balducci I, Koga CY. Comparaçao da actividade antimicrobiana de soluçoes de peróxido de hidrógeno e malva sobre candida albicans. Cienc Odontol Bras 2009 abr/jun.;12(2).24-28.
-
Estela V. Kloc. Farmacodinamia de la Clorhexidina y su aplicación en odontopediatria. Boletín de la Asociación Argentina de Odontología para niños. Dic2003/marzo2004. Vol. 32(4): 17-19.

