










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Medicina Interna
Print version ISSN 0250-3816On-line version ISSN 1688-423X
Arch. Med Int vol.37 no. 3 Montevideo Nov. 2015
ARCHIVOS DEL INSTITUTO DE NEUROLOGÍA
Caso Clínico de Interés
Primera experiencia de tratamiento con everolimus en un paciente con esclerosis tuberosa y astrocitoma subependimario de células gigantes en Uruguay
First experience of everolimus in patients with tuberous sclerosis and subependymal giant cell astrocytoma in Uruguay
Dr. Diego Costales
Residente de Neuropediatría. Cátedra de Neuropediatría. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Dr. Conrado Medici
Profesor Adjunto de Neuropediatría. Cátedra de Neuropediatría. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Dr. Alfredo Cerisola
Profesor Agregado de Neuropediatría. Cátedra de Neuropediatría. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Dr. Gonzalo Costa
Profesor Agregado de Neurocirugía. Cátedra de Neurocirugía. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Dr. Emilio Turcati
Imagenólogo. Colaborador de la Cátedra de Neuropediatría. Cátedra de Neuropediatría. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Dr. Gustavo Dufort
Coordinador del Centro Hemato-Oncólogo Pediátrico. Centro de Hemato-Oncológico Pediátrico. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Dr. Gabriel Gonzalez
Profesor de Neuropediatría. Cátedra de Neuropediatría. Centro Hospitalario Pereira Rossell. Montevideo, Uruguay
Recibido: 7/9/15 - Aceptado: 13/09/15
Institución responsable: Centro Hospitalario Pereira Rossell.
Correspondencia: Dr. Conrado Medici. Bulevar Artigas 1550. Montevideo, Uruguay. E-mail: conrado.medici@gmail.com
Resumen: Arch Med Interna 37(3):
El complejo esclerosis tuberosa (CET) es una enfermedad genética multisistémica determinada por la pérdida de función del complejo hamartina/tuberina. Esto causa activación del m-TOR con formación de tumores como el astrocitoma subependimario de células gigantes (SEGA). El everolimus inhibe el m-TOR reduciendo el tamaño de los SEGA. El objetivo de este trabajo es mostrar la experiencia del primer caso de SEGA tratado con everolimus en Uruguay. Caso: Escolar de 6 años, con desarrollo normal, sin epilepsia y portador de CET. A los 5 años instala hipertensión intracraneana por hidrocefalia secundaria al SEGA. Se realiza cirugía lográndose resección parcial, con remanente tumoral de 14.3cm3. Se inició everolimus evidenciando las resonancias magnéticas de control una reducción del tamaño tumoral del 54.5% al mes, del 65.5% a los 3 meses y del 68.5% a los 6 meses de iniciado el tratamiento. Presentó efectos secundarios menores (aftas bucales, infecciones respiratorias altas). La buena respuesta obtenida aporta evidencia del uso del everolimus como tratamiento complementario a la cirugía en los pacientes con SEGA.
Palabras claves: Esclerosis Tuberosa, Astrocitoma, Everolimus
Abstract: Arch Med Interna 37(3):
Tuber sclerosis complex (TSC) is a multisystemic genetic disease caused by a loss of function of hamartin/tuberin complex. This results in an m-TOR activation with tumor formation, among which is subependymal giant cell astrocytoma (SEGA). Everolimus inhibits m-TOR reducing SEGA size. The objective of this work is to show the first experience of SEGA treatment with everolimus in Uruguay. Case: 6 years old boy with TSC, with normal development and no history of seizures. At the age of 5 he installed intracranial hypertension due to hydrocephalus secondary to a SEGA. With surgery tumor was partially resect, with a 14.3cm3 tumor remnant. Treatment with everolimus was iniciated with a tumor size reduction of 54.5% after the first month of treatment, 65.5% after 3 months and 68.5% after 6 months. He had minor secondary effects (mouth ulcerations and upper respiratory tract infections). Good response in this patient shows evidence of possible use of everolimus as an adjuvant terapy to surgery in SEGA.
Key words: Tuber Sclerosis, Astrocytoma, Everolimus
Introducción
El complejo esclerosis tuberosa (CET) es una enfermedad genética multisistémica autosómica dominante, de expresión variable (1,2). Es causada por la mutación inactivante de uno de los genes TSC1 o TSC2 (1-4). El gen TSC1 se ubica en el cromosoma 9q34 y codifica para la proteína hamartina y el gen TSC2 se ubica en el cromosoma 16p13.3 y codifica la proteína tuberina (1-4). Estas proteínas conforman el complejo hamartina/tuberina, un supresor tumoral que inhibe la proteína mammalian target of rapamycin (m-TOR) la que promueve la proliferación, el crecimiento y el metabolismo celular (2,5). La pérdida de función de TSC1 o TSC2 causa activación constitucional de la m-TOR causando la formación de tumores hamartomatosos en diferentes topografías, como el astrocitoma subependimario de células gigantes (SEGA) (5,6).
La incidencia del CET es de 1/6000 a 1/10000 recién nacidos vivos con una prevalencia en la población general de 1/20000, y de 1/12000 a 1/14000 en niños menores de 10 años (1,2,7).
El CET puede presentar compromiso variable del sistema nervioso central (SNC), piel, ojos, corazón, riñones y pulmón (2). Estos órganos se afectan en mayor o menor medida en los diferentes pacientes, apareciendo algunas lesiones a determinadas edades (2). Un 85% de los niños y adolescentes con CET presentan manifestaciones del sistema nervioso central, incluyendo epilepsia, discapacidad intelectual, dificultades de aprendizaje, trastorno por déficit atencional e hiperactividad, trastornos de conducta y trastorno del espectro autista (1,2).
Los primeros criterios diagnósticos de CET fueron descriptos por Gómez en 1979 (7). En 1998 se desarrolló el primer consenso internacional que definió los criterios diagnósticos (8), los cuales se actualizaron mediante un nuevo consenso en 2012 (9). A diferencia del primer consenso, el segundo define criterios diagnósticos genéticos y criterios diagnósticos clínicos mayores y menores. Los criterios diagnósticos genéticos son la identificación de mutaciones patogénicas en los genes TSC1 o TSC2. La presencia de dichas mutaciones es suficiente para hacer diagnóstico definitivo de CET, pero en un 10-25% de los pacientes no se identifica mutación (9). En ese mismo consenso se definieron criterios de seguimiento y manejo de los pacientes con CET (10), y de diagnósticos, de screening y tratamiento para el SEGA (11).
Dentro del SNC se incluyen como criterios diagnósticos mayores la presencia de displasias corticales (túberes y/o líneas de migración neuronal en la sustancia blanca), nódulos subependimarios o el SEGA (9). Los SEGA se definen como lesiones tumorales en la región de la ranura caudotalámica con un tamaño mayor a 1cm en cualquier dirección o lesiones subependimarias en cualquier localización que han demostrado crecimiento en las imágenes consecutivas sin importar el tamaño de las mismas (11). En general los SEGA captan ávidamente el contraste pero cualquier lesión subependimaria en crecimiento aunque no capte contraste debe ser considerada SEGA (11). Su prevalencia se ha estimado hasta en un 5-20% de los pacientes con CET (5,11,12). Se plantea que se desarrollan a partir de un nódulo subependimario, en general son de lento crecimiento y al ubicarse a nivel del foramen de Monro pueden bloquear la circulación del líquido cefalorraquídeo y producir hidrocefalia (1).
El tratamiento del SEGA históricamente ha sido solamente quirúrgico dado la falta de respuesta de estas lesiones a la quimioterapia y a la radioterapia y a la posible malignización secundaria al usar estos tratamientos (11,13). En el año 2006 Franz y colaboradores describieron que la rapamicina, un inhibidor del m-TOR, era eficaz en el tratamiento de los SEGA, produciendo remisión de los mismos (5). Luego se vio que el everolimus, otro inhibidor del m-TOR, derivado de la rapamicina, era eficaz y seguro para el tratamiento del SEGA (14). A partir de esa fecha varios trabajos se han realizado intentando mostrar la eficacia y la seguridad de los inhibidores del m-TOR en el tratamiento del SEGA. En el consenso del 2012 se incluyeron como una opción terapéutica a la cirugía en los SEGA asintomáticos, en crecimiento, frente a la presencia de una enfermedad multisistémica, de SEGA múltiples o infiltrantes o en los casos en que está contraindicada la cirugía o en que se plantea que en la misma no se va a obtener una resección completa (11).
El objetivo de esta publicación es mostrar la experiencia del primer caso de SEGA tratado con everolimus en Uruguay.
Caso clínico
Escolar de 6 años, con desarrollo normal, sin epilepsia y portador de CET diagnosticado a los 14 meses por presentar más de 3 máculas hipopigmentadas, imágenes compatibles con túberes corticales en la resonancia magnética (RM) de cráneo y rabdomioma cardíaco. No se evidenció un SEGA al momento del diagnóstico ni en el control imagenológico realizado a los 2 años y 7 meses. Luego de eso no se realizaron nuevos controles imagenológicos.
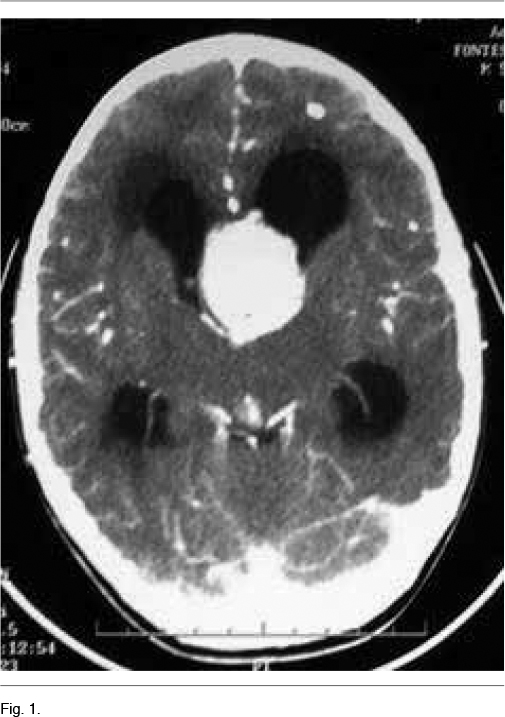
A los 5 años instala un síndrome de hipertensión endocraneana dado por cefaleas que progresan en el correr de tres meses, llegando los días previos a la consulta a despertarlo en la noche y a acompañarse de vómitos matinales que alivian las cefaleas. La tomografía computada de cráneo de urgencia informa un proceso expansivo de tercer ventrículo, que obstruye el agujero de Monro, determinando una hidrocefalia supratentorial de ambos ventrículos laterales, mayor a izquierda, con desviación de línea media a derecha de 12mm, edema transependimario e intensa captación de contraste (Figura 1). Con planteo de hidrocefalia secundaria a SEGA, se realiza en una primera instancia una derivación ventrículo-peritoneal de urgencia a nivel del ventrículo lateral derecho, para tratamiento de su hipertensión endocraneana descompensada. El procedimiento no presenta complicaciones, quedando el paciente asintomático luego del mismo. En el fondo de ojo post-derivación se observa edema de papila bilateral. La RM post-operatoria muestra el SEGA y la persistencia de una hidrocefalia a nivel del ventrículo lateral izquierdo. En una segunda instancia se realiza la cirugía del tumor lográndose una resección parcial del mismo, con un remanente tumoral de 14.3cm3 (Figura 2A) La anatomía patológica confirmó el diagnóstico de SEGA.
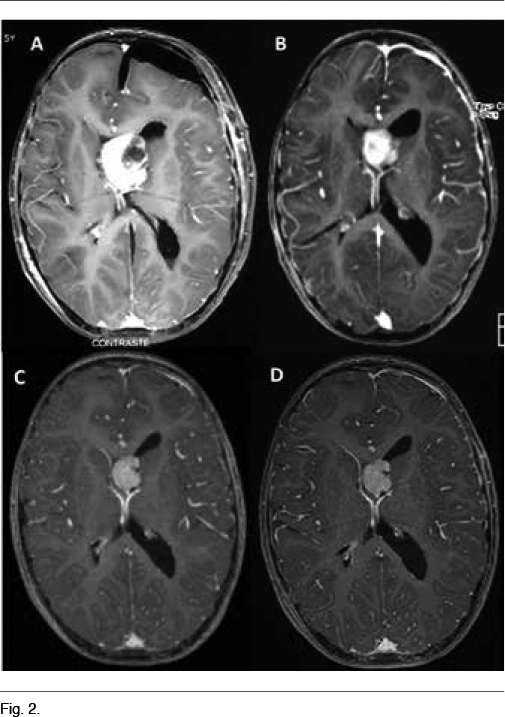
Se decide iniciar everolimus, a pesar que su indicación como tratamiento adyuvante es off-label (11). Se inició everolimus a 2.5mg/día (3mg/m2/día) aumentándose la dosis a 5 mg/día por presentar dosificación sérica de 1.5ng/ml, por debajo del rango terapéutico (3-15ng/ml), lográndose una concentración de 9.4ng/ml. En las RM de control se evidenció una reducción del tamaño tumoral del 54.5% al mes (6.5cm3) (Figura 2B), del 65.5% a los 3 meses (4.9cm3) (Figura 2C) y del 68.5% a los 6 meses (4.5cm3) (Figura 2D). Como efectos secundarios presentó astenia al inicio del tratamiento, aftas bucales que no interfirieron con la alimentación ni requirieron tratamiento y 3 episodios de infección respiratoria alta que fueron bien toleradas y no requirieron modificaciones en la medicación. Los controles paraclínicos no evidenciaron alteración en la función renal, perfil lipídico, glicemia, ionograma, funcional y enzimograma hepático, en el hemograma ni en la orina.
El paciente se mantiene asintomático desde el punto de vista neurológico y el fondo de ojo de control es normal.
Discusión
Presentamos el primer caso de un niño con CET y SEGA tratado en Uruguay con everolimus en el cual se obtuvo una muy buena respuesta con importante reducción del tamaño tumoral y buena tolerancia al tratamiento.
Se trata de un niño de 6 años que tiene la particularidad de no presentar epilepsia ni retraso del desarrollo. La epilepsia está presente en aproximadamente un 85% de los pacientes con CET, iniciando generalmente en el primer año de vida, pudiendo presentarse como un Síndrome de West y ser fármacoresistente (2). Hasta un 50% de los pacientes pueden presentar discapacidad intelectual, y los niños con nivel intelectual promedio es frecuente que tengan déficit atencional, trastornos mnésicos y que sean disejecutivos (2).
En este paciente el SEGA no estaba presente a los 18 meses ni a los 2 años y 7 meses. Luego presentó un rápido crecimiento con desarrollo de hidrocefalia e hipertensión endocraneana. En general los SEGA son tumores de lento crecimiento (1). En los pacientes con CET sin SEGA se recomienda realizar una RM de control cada 1 a 3 años, independientemente que el paciente este asintomático, hasta los 25 años, fecha a partir de la cual se sabe que excepcionalmente se va a desarrollar un nuevo SEGA (10,11). Frente a la presencia de una SEGA que no está en crecimiento, en un paciente asintomático, se recomienda realizar RM cada 6 meses a 1 año. La neuroimágen se debe adelantar frente a la presencia de nueva sintomatología como cefaleas, alteración visual, nauseas, vómitos, aumento del perímetro cefálico o frente a un aumento del número de crisis en un paciente epiléptico (11).
En nuestro paciente se optó por tratamiento quirúrgico en primera instancia, si bien a priori el volumen tumoral hacía difícil la posibilidad de una resección tumoral completa. Dado la presencia del remante tumoral post-quirúrgico y el riesgo de posibles secuelas neurológicas frente a una nueva intervención, se decidió iniciar tratamiento con everolimus. El mismo fue indicado off-label ya que hasta la fecha no se ha aprobado el uso de los inhibidores del m-TOR para el tratamiento post-quirúrgico de los pacientes con SEGA y remanente tumoral (11).
El consenso internacional del año 2012 define que los SEGA sintomáticos o aquellos asociados a aumento del tamaño ventricular, cambios inexplicados en el estado neurológico o síntomas de trastornos neuropsiquiatricos asociados a CET requieren intervención quirúrgica o monitoreo clínico y realización de una nueva imagen (11). En los SEGA con síntomas agudos recomienda que la intervención sea quirúrgica (11). En los SEGA asintomáticos pero que están en crecimiento se recomienda resección quirúrgica o inhibidores de la m-TOR. La decisión dependerá del riesgo de complicaciones, efectos adversos, costos, duración del tratamiento y presencia de comorbilidades (11). Los SEGA únicos, unilaterales, totalmente resecables sin factores de riesgo u otras comorbilidades se beneficiarán de cirugía (11). Pacientes con enfermedad multisistémica o con SEGA múltiples o infiltrantes, que no se plantea la resección completa o si hay contraindicación para la cirugía se beneficiarán de inhibidores de la m-TOR (10,11).
Los inhibidores de la m-TOR imitan la acción inhibitoria del complejo hamartina/tuberina sobre la m-TOR lo que evita el exceso de proliferación y crecimiento celulares que ocurre tras la pérdida de función de este complejo. En los pacientes con CET se ha demostrado que el uso de everolimus lleva a una reducción del tamaño de los SEGA (15).
Los estudios actuales sobre everolimus en tratamiento de SEGA han demostrado que es eficaz con reducciones del volumen tumoral del 50% o mayor en el 35% de los pacientes tratados vs 0% pacientes con placebo (p<0.0001). Es un tratamiento seguro con efectos adversos leves frecuentes (infecciones respiratorias altas, aftas orales) pero con efectos adversos severos poco frecuentes. Como efecto adicional se encontró que hubo disminución significativa o estabilización en los angiomiolipomas que asociaban los pacientes (15). El everolimus es también eficaz en el tratamiento de los angiomiolipomas renales y se ha descripto que produce reducción del número y tamaño de los nódulos subependimarios, de los rabdomiomas cardíacos, y que podría disminuír la frecuencia de crisis epilépticas en los pacientes con epilepsia, aunque faltan investigaciones en esta línea (16). El tratamiento a largo plazo demostró una reducción sostenida de los SEGA siendo mayor en los primeros meses de tratamiento y fue bien tolerado, no encontrándose aumento significativo de los efectos adversos (16,17). Aún falta por definir por cuánto tiempo puede ser necesario continuar el tratamiento y los posibles efectos adversos del tratamiento a largo plazo.
En conclusión mostramos la buena respuesta de nuestro paciente al tratamiento del SEGA con everolimus, con escasos efectos adversos que no implicaron una suspensión del mismo, lo que coincide con lo descripto en la literatura. La buena respuesta obtenida en este paciente aporta evidencia de su posible uso como tratamiento adyuvante a la cirugía en los pacientes con SEGA.
Bibliografía
1. Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657-68.
2. Curatolo P, Maria BL. Tuberous sclerosis. Handb Clin Neurol. 2013; 111:323-31.
3. The European chromosome 16 Tuber sclerosis consortium. Identification and characterization of the Tuberous sclerosis gene on chromosome 16. Cell 1993;75:1305-15.
4. van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997;277:805-8.
5. Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490-8 .
6. Budde K, Gaedeke J. Tuberous sclerosis complex-associated angiomyolipomas: focus on mTOR Inhibition. Am J Kidney Dis. 2012;59:276–83
7. Sampson JR, Scahill SJ, Stephenson JB, Mann L, Connor JM. Genetic aspects of tuberous sclerosis in the west of Scotland. J Med Genet.1989;26:28-31.
8. Roach ES, Gómez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria J Child Neurol. 1998;13:624-8 .
9. Northrup H, Krueger DA, Roberds S, Smith K, Sampson J, Korf B, et al. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49:243-54.
10. Krueger DA, Northrup H, Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49:255-65
11. Roth J, Roach ES, Bartels U, Jóźwiak S, Koenig MK, Weiner HL, et al. Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous sclerosis complex consensus conference 2012. Pediatr Neurol. 2013;49:439-44.
12. Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol. 2009;16:691-6
13. Matsumura H, Takimoto H, Shimada N, Hirata M, Ohnishi T, Hayakawa T. Glioblastoma following radiotherapy in a patient with tuberous sclerosis. Neurol Med Chir (Tokyo). 1998;38:287-91.
14. Krueger D, Care M, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant cell astrocytoma in Tuberous sclerosis. N Eng J Med. 2012;363:1801-11.
15. Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–32.
16. Tran LH, Zupanc ML. Long-term everolimus treatment in individuals with Tuberous sclerosis complex: a review of the current literatura. Pediatr Neurol. 2015;53:23-30.
17. Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Everolimus for subependymal giant cell astrocytoma in patient with tuberous sclerosis complex: 2 year open-label extensión of the randomised EXIST-1 study. Lancet Oncol. 2014;15:1513-20.

