










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Medicina Interna
versión impresa ISSN 0250-3816versión On-line ISSN 1688-423X
Arch. Med Int vol.36 no. 3 Montevideo nov. 2014
Artículo Original
Citopenias hematológicas en enfermedades autoinmunes sistémicas
Blood cytopenias in systemic autoimmune conditions
Dra. Luisa Servioli
Internista. Reumatóloga. Ex Asistente Clínica Médica 1. Facultad de Medicina. UdelaR. Montevideo.
Dr. Jorge Facal
Profesor Director Clínica Médica 1. Facultad de Medicina. UdelaR. Montevideo.
Dra. Sandra Consani
Internista. Profesora Adjunta Clínica Médica 3. Facultad de Medicina. UdelaR. Montevideo.
Dr. Gabriel Maciel
Internista. Reumatólogo. Profesor Agregado Clínica Médica 1. Facultad de Medicina. UdelaR. Montevideo.
Dr. Alejandro Fernández
Internista. Asistente Clínica Médica 1. Facultad de Medicina. UdelaR. Montevideo.
Recibido: 11/06/14 - Aceptado: 03/10/14
Trabajo de la Policlínica de Enfermedades Autoinmunes Sistémicas. Departamento de Medicina. Hospital Maciel. Facultad de Medicina. UdelaR. Montevideo. Uruguay.
Correspondencia: Dra. Luisa Servioli. Policlínica de Enfermedades Autoinmunes. Sector Policlínicas, Hospital Maciel, 25 de Mayo 174, Montevideo, Uruguay. Tel: (598) 2915 3000, interno 2126. E-mail: lfservioli@hotmail.com
Resumen:
Las citopenias hematológicas son un hallazgo frecuente y potencialmente grave en las Enfermedades Autoinmunes Sistémicas. Sus causas pueden ser muy variadas, de ahí la importancia de un abordaje diagnóstico sistematizado que asegure además el tratamiento correcto. En este artículo se revisan las generalidades de las citopenias hematológicas en cuanto a frecuencia, causas, así como su enfoque diagnóstico y terapéutico.
Palabras clave: citopenias inmunomediadas, citopenias hematológicas en enfermedades autoinmunes, anemia inflamatoria, leucopenia autoinmune, neutropenia en enfermedades autoinmunes.
Abstract:
The hematologic cytopenias are a frequent and potentially dangerous finding in the Systemic Autoimmune Diseases. It may have different etiologies, and for that reason it is important a systematic approach to ensure the correct diagnosis and treatment. In this article, the frequency, etiology, diagnostic approach and treatment of the hematologic cytopenias are reviewed.
Keywords: autoimmune cytopenias, hematologic cytopenias in autoimmune diseases, inflammatory anemia, autoimmune leukopenia, neutropenia in autoimmune diseases.
Introducción
Las citopenias hematológicas son una complicación frecuente y potencialmente grave en las Enfermedades Autoinmunes Sistémicas (EASs). Sus causas pueden ser muy variadas y con tratamientos incluso contrapuestos. De ahí la importancia de un abordaje sistematizado de las mismas, que asegure el diagnóstico etiológico correcto, el tratamiento adecuado y una óptima utilización de los recursos disponibles. Este artículo trata sobre las generalidades de las citopenias hematológicas en las EASs, su frecuencia, causas y mecanismos patogénicos, proponiéndose algoritmos diagnósticos y realizando una breve reseña sobre el tratamiento.
La mayoría de los estudios sobre el tema se han realizado en pacientes con Lupus Eritematoso Sistémico (LES). En las EASs distintas del LES, la información disponible se basa en series pequeñas y reporte de casos. Sin embargo, varios de los conceptos que se mencionan para el LES pueden generalizarse a las otras EASs.
Tipos de citopenias hematológicas en el LES
Las hemocitopenias están incluídas en los criterios de clasificación para el LES, tanto del American College of Rheumatology (1997)(1) (Tabla I) como en los del Systemic Lupus International Collaboratory Clinics Group (2012)(2), lo que evidencia lo frecuente que es el compromiso hematológico en esta enfermedad.
La Tabla II muestra las frecuencias de las citopenias hematológicas reportadas por las distintas series en pacientes con LES. La anemia es la alteración hematológica más frecuente(3,4) (50-80% de los casos), seguida de cerca por la linfopenia.
La anemia es también la manifestación hematológica más común en las otras EASs.
Causas y mecanismos patogénicos de las hemocitopenias en pacientes con LES.
Las causas más frecuentes de citopenias hematológicas en el LES son inmunológica, farmacológica, infecciosa, carencias nutricionales e insuficiencia renal crónica (IRC) (estas dos últimas vinculadas sobre todo con anemia).(5,6) Otras causas menos frecuentes son mielodisplasia, neoplasia, CID y PTT.(5,6) Estas causas son similares a las descritas para el resto de las EASs.


Causa inmunológica
En estos casos la citopenia constituye una manifestación de la enfermedad autoinmune de base. La causa autoinmune suele ser un diagnóstico de exclusión. Los mecanismos patogénicos planteados para las hemocitopenias de causa inmunológica son: 1) destrucción periférica de células sanguíneas mediada por anticuerpos y 2) daño inmunológico primario de la médula ósea (MO).(5,6)
Destrucción periférica de células sanguíneas
La destrucción periférica de células sanguíneas es el principal mecanismo patogénico de la linfopenia, neutropenia y trombocitopenia en el LES.
Está mediado por anticuerpos dirigidos contra la célula sanguínea propiamente dicha, sus precursores, factores de crecimiento o receptores de dichos factores.
Se han implicado distintos mecanismos en las diferentes citopenias que se pasan a analizar por separado en cada caso.
En la linfopenia: a) Anticuerpos linfocitotóxicos: Son frecuentes en las EASs, sobre todo en el LES (36-90%).(6-9,10) Su significado patogénico es incierto y dudosa la importancia de su determinación en la práctica clínica.(6) b) Déficit en la expresión linfocitaria de proteínas de superficie reguladoras del complemento (CD55 y CD59), con mayor susceptibilidad a la lisis mediada por complemento.(6,11) c) Producción endógena de IFNs tipo 1 e IFNα.(6,12,13)
En la neutropenia: a) Anticuerpos antineutrófilo.(6,14) b) Anticuerpos contra precursores mieloides, anti-G-CSF y resistencia de células mieloides al G-CSF (que podría estar mediado por anticuerpos).(6,10,15,16) c) Apoptosis neutrófila incrementada inducida por TNF.(6)
En la trombocitopenia: a) Destrucción periférica de plaquetas mediada por anticuerpos antiplaquetarios. Es el mecanismo patogénico más común de trombocitopenia en el LES.(6, 17-19) b) Otros anticuerpos implicados son los antifosfolípido (AAF) (6, 20), antitrombopoyetina (TPO) (6, 19, 21) y antireceptor de la TPO.(6)
Anemia Hemolítica Autoinmune (AHA): a) Anticuerpos calientes de tipo IgG (ocasionalmente IgA). b) Otros anticuerpos como anticuerpos anticardiolipina (aCL) o anticoagulante lúpico (AL), existen varios estudios de pacientes con LES que señalan una correlación significativa entre los mismos y anemia hemolítica Coombs positiva.(5) Los aCL podrían contribuir en la patogenia de la AHA actuando como anticuerpos anti- eritrocitos.(5) La hemólisis autoinmune parece ser marcador de un subgrupo de pacientes con LES con mayor prevalencia de Síndrome Antifosfolípido.(5)
Anemia inflamatoria: existe una producción disminuida de eritropoyetina (EPO)(22) y resistencia primaria a su acción.(5, 23-25) Ambos eventos son atribuidos a las citoquinas inflamatorias, como TNF α, IL1 β, IL6, INF γ (22- 25) y a la presencia de anticuerpos antiEPO.(5, 25- 27)
Daño inmunológico primario de la MO
Las citopenias autoinmunes tendrían un componente central, resultado del daño inmunológico primario de la MO, que sería un órgano blanco en las EASs.(5,6)
Los autoanticuerpos, linfocitos T y la disregulación de citoquinas provocarían distintos tipos de falla hemopoyética inmunomediada, determinando desde alteraciones morfológicas hasta entidades bien definidas de insuficiencia medular como anemia aplásica, hipoplasia de la línea mieloide, trombocitopenia amegacariocítica y aplasia pura de células rojas (APCR)(5,28).
La evidencia de daño inmunológico primario de la MO surge de:
- Los hallazgos en la BMO de pacientes con LES y citopenias, de hipocelularidad y displasia de las 3 series, fibrosis, necrosis de la MO e infiltración linfoplasmocitaria(5,6,29,30). La displasia a veces es transitoria, relacionada con actividad de la EAS y desaparece con la remisión.(31)
- El hallazgo en pacientes con LES y anemia aplásica (insuficiencia medular de probada patogenia autoinmune) de autoanticuerpos séricos inhibitorios de la hematopoyesis.(5) Estos anticuerpos se relacionan con la actividad del LES y pueden ser inhibidos mediante inmunosupresores (IS) o plasmaféresis.
- Supresión de la hemopoyesis mediada por linfocitos T autoreactivos. Sería la principal causa de falla de la MO en pacientes con LES.(5)
- Depleción apoptótica de las stem cells de la MO por expresión incrementada del gen Fas, atribuida al aumento de TNF α, INF γ, y linfocitos T citotóxicos de la MO.(5)
- Insuficiencia del estroma de la MO y producción disminuida de factores de crecimiento hemopoyético(5).
Causa farmacológica
Es una de las causas más frecuentes de hemocitopenias. La suspensión o reducción de la dosis del fármaco sospechoso constituye una prueba diagnóstica y es una de las primeras medidas e incluso, en ocasiones, podría ser la única conducta a tomar. Sin embargo, siempre deben valorarse las otras posibles causas de citopenias.
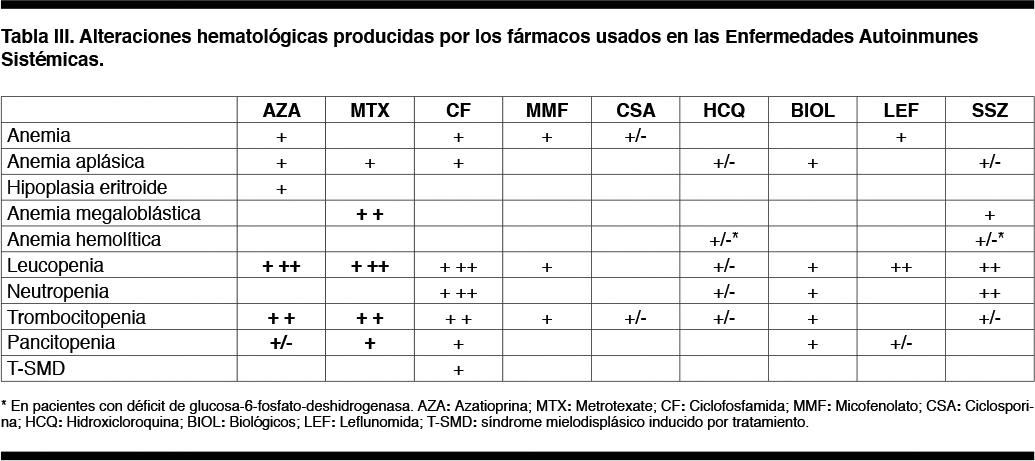
La Tabla III muestra las alteraciones hematológicas más frecuentes producidas por los fármacos usados en el tratamiento de las EASs. Los que más comúnmente causan citopenias por su acción mielotóxica son la Azatioprina (AZA), Metrotexate (MTX) y Ciclofosfamida (CF). Suele ser una mielosupresión reversible, generalmente leve (aunque puede ser grave) y dependiente de la dosis. La toxicidad medular por MTX responde a la administración de folatos.(32-34)
La leucopenia y trombocitopenia son los hallazgos más comunes.(6) La leucopenia y neutropenia por CF son máximas a los 8-12 días después de la administración intravenosa. Debe evitarse que el recuento leucocitario descienda a menos de 3.000/mm3 y que los neutrófilos sean inferiores a 1.000/mm3 ajustando la dosis hasta alcanzar los niveles deseados.(32)
La mielotoxicidad es menos común con el uso de Micofenolato (MMF), Ciclosporina (CSA) e Hidroxicloroquina. Los hallazgos específicos en la MO pueden sugerir mielotoxicidad inducida por drogas.
La neutropenia también puede aparecer con el tratamiento con Tocilizumab y Rituximab, mientras que son infrecuentes las citopenias durante el tratamiento con otros agentes biológicos.
Son escasos los reportes sobre síndromes mielodisplásicos (SMD) provocados por el tratamiento de las EASs, estando vinculados sobre todo con la CF. En estos casos, el examen de MO es diagnóstico por las severas alteraciones morfológicas y citogenéticas, presentando un riesgo elevado de transformación leucémica.(35)
De los estados relacionados a la panmielosis inducidas por tratamiento, los más precoces podrían ser reversibles al retirar rápidamente el fármaco.
Infecciones
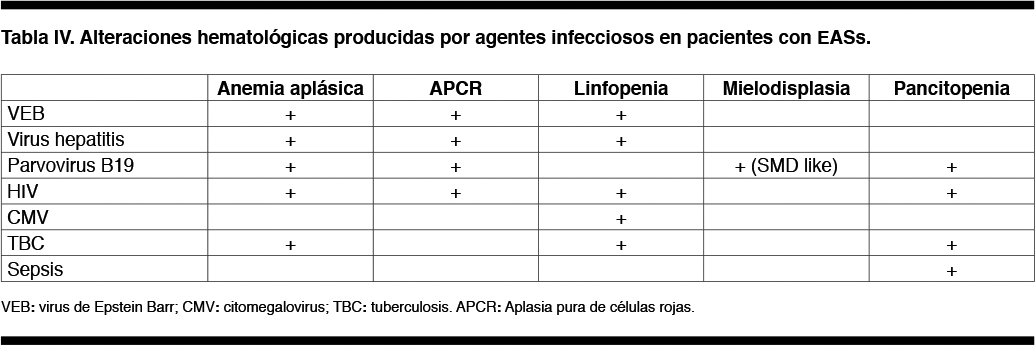
Las infecciones deben considerarse como causa de citopenias hematológicas en pacientes con EASs, más aún si el paciente cursa con fiebre.
La Tabla IV muestra la relación entre los microorganismos más frecuentes (sobre todo virus) y diversas alteraciones hematológicas en pacientes con EASs. Por ejemplo, el Parvovirus B19 provoca cambios “SMD” like transitorios en la MO o crisis aplásicas transitorias, con mejoría espontánea sin tratamiento.(10,35)
La tuberculosis diseminada y la sepsis son causa de pancitopenia en los pacientes inmunodeprimidos.
Neoplasias
Pueden ser causa de citopenias porque los pacientes con EASs tienen un riesgo aumentado de desarrollar neoplasias hematológicas, sobre todo síndromes linfoproliferativos malignos, como el LNH. Esta asociación se ha reportado en el Síndrome de Sjögren Primario (SSp), (que presenta la más alta incidencia de LNH dentro de las EASs), LES, Artritis Reumatoide (AR), y Polimiositis-Dermatomiositis.(36-39)
Por otro lado, las EASs también pueden ser expresión de síndromes paraneoplásicos de tumores hematológicos y de algunos tumores sólidos.
Síndromes mielodisplásicos
La relación entre EASs y los SMD es controvertida aunque se ha hallado asociación entre estas entidades. Se ha propuesto un componente autoinmune en la patogenia de los SMD primarios. Esto se basa en la observación de que algunos pacientes con SMD presentan manifestaciones autoinmunes (22%),(10,40) disregulación de la inmunidad humoral/celular (10,41) y buena respuesta a la terapia inmunosupresora.(10)
A su vez se han reportado casos de EASs (SSp, Polimialgia Reumática, LES, Enfermedad Mixta del Tejido Conectivo , AR) complicadas con SMD, sin mediar agentes mutagénicos o radiación, sugiriendo que la causa puede ser la disfunción inmune hallada en las EASs.(41,42) El uso de tratamiento inmunosupresor en los SMD es discutido, a pesar de este planteo patogénico, por el aumento de riesgo de transformación leucémica.
ANEMIA EN PACIENTES CON LES
Causas de anemia en el LES
Si bien el espectro etiológico es amplio (Tabla V), las causas que explican la mayoría de los casos de anemia en el LES son:
Anemia inflamatoria crónica. Es la más común (37-66,7%).(5,25) Se correlaciona con actividad de la enfermedad. La severidad de la anemia es leve a moderada y suele coexistir con anemia causada por otros mecanismos.(5) Este mecanismo de anemia (enfermedades crónicas) también es el más frecuente en las otras EASs.
Anemia carencial ferropénica. Por pérdidas sanguíneas digestivas (debido al uso de corticoides, AINEs, AAS, anticoagulantes) o menorragia (dado lo común de las alteraciones menstruales).(5)
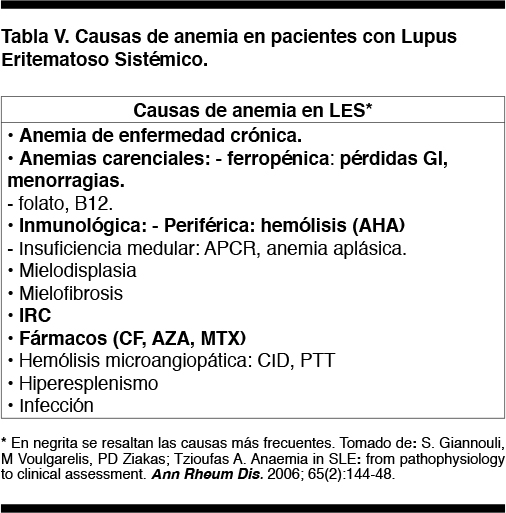
AHA
Anemia por IRC
Mielotoxicidad inducida por drogas (CF, AZA y MTX).(5)
Características de la anemia en el LES
El estudio de Voulgarelis y cols. resume las características generales de la anemia en pacientes con LES. (25) Este estudio prospectivo, de 3 años de duración, evaluó la prevalencia de las diferentes causas de anemia y su asociación con distintos parámetros clínicos e inmunológicos en 132 pacientes con LES.
La prevalencia de anemia en el LES fue de 38%. Las causas identificadas se dividieron en 4 grupos: anemia inflamatoria crónica 37,1%, anemia ferropénica 35,6% (la mayoría mujeres con pérdidas menstruales incrementadas), AHA 14,4% y otras causas 12,9% (la mayoría eran anemias por IRC y menos común por β talasemias, mielotoxicidad por CF, PTT o CID).
La severidad de la anemia fue globalmente leve (HB 9-11 g/dl). Sin embargo, se observó una variación significativa en la severidad de la anemia según la causa (p < 0,001), correspondiendo los casos más graves a AHA. Los 4 grupos difirieron en el score de actividad del LES, siendo el grupo con anemia inflamatoria el de mayor actividad. En cuanto a la evolución de la anemia, la AHA remitió rápidamente (media de tiempo de corrección 3 meses) y la tasa de recurrencia fue baja. La anemia ferropénica llevó más tiempo en corregir (media 10 meses), según los autores por demora en el tratamiento. Todos los pacientes corrigieron la anemia en estos 2 grupos. La remisión fue infrecuente en la anemia inflamatoria, más de la mitad de los pacientes permanecieron con anemia durante el seguimiento de 3 años. La recuperación en la anemia inflamatoria fue particularmente lenta en pacientes que recibían IS. Se desconoce si el uso de IS es la causa directa de la supresión persistente de la MO o si esta es un marcador de actividad de la enfermedad que requiere el uso de los mismos.(25) En relación a las otras causas de anemia se señala que tuvieron un comportamiento mixto, la mitad remitieron en tiempos similares a las ferropénicas, mientras que el resto no corrigieron por largos períodos de tiempo.(25)
Algoritmos diagnósticos para la anemia en pacientes con LES
En la orientación etiológica inicial de un paciente con anemia, es fundamental la historia clínica y examen físico, junto a la realización de sencillos tests de laboratorio, que permitan tipificar la anemia (severidad, si es pura o no, VCM, reticulocitosis, etc.).
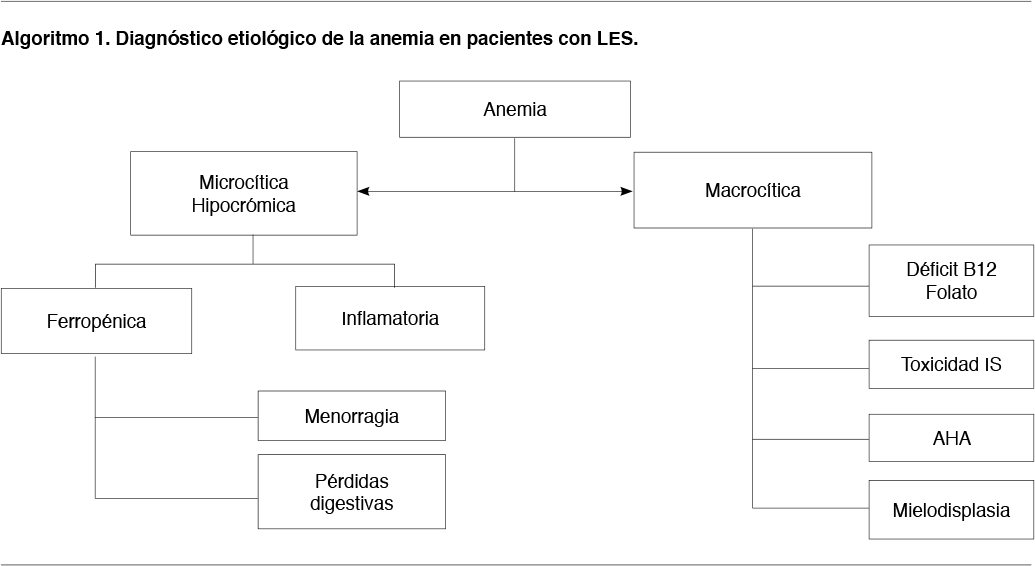
Anemia microcítica hipocrómica (Algoritmo 1)
La etiología más probable es la carencial ferropénica. En ese caso la menorragia o pérdidas digestivas son las causas más probables, y si la historia es compatible se deben dirigir los estudios en ese sentido. Se debe recordar al analizar el metabolismo del hierro, que la ferritina es un reactante de fase aguda y que en estos casos puede estar disminuída en menor grado ya que subyace también un mecanismo asociado a las enfermedades inflamatorias crónicas. Otra causa de ferropenia, aunque menos frecuente, es la enfermedad celíaca, que puede asociarse a las EASs. Ante esta sospecha diagnóstica, los anticuerpos antitransglutaminasa pueden usarse como screening. Descartada la ferropenia, la anemia microcítica hipocrómica puede corresponder a anemia de las enfermedades crónicas (1/3 de los casos).
Anemia macrocítica (Algoritmo 1)
Puede ser carencial por déficit de folato o de vitamina B 12 (aunque la anemia perniciosa es rara), o deberse a toxicidad por IS (MTX).
Otras causas pueden ser AHA con consumo de ácido fólico o la mielodisplasia.
Anemia normocítica-normocrómica (NN). (Algoritmo 2)
La causa más probable, si es regenerativa, es la AHA. La hemólisis debe confirmarse y buscar asociación lesional con PTA y AAF. En anemias NN y macrocíticas siempre conviene descartar un componente hemolítico subclínico.
Si es arregenerativa, es fundamental la orientación según la clínica del paciente. De ser severa y/o persistente o si se presenta en el contexto de bicitopenia o pancitopenia debe realizarse mielograma y eventual BMO. El estudio de MO podrá evidenciar en estos casos insuficiencia medular. Se debe valorar si la causa es mielotoxicidad por fármacos (los hallazgos en la MO pueden sugerir este diagnóstico), si la falla puede ser secundaria a infección (el diagnóstico puede sospecharse por la clínica, fiebre, mielocultivo y serologías) o si se trata de una sustitución medular. Una vez excluidas estas causas la insuficiencia medular podría ser de causa inmunológica.
Si la anemia arregenerativa es leve/moderada y pura, lo más probable es que se trate de una anemia inflamatoria crónica. Es un diagnóstico de exclusión por lo que se deben descartar otras causas de anemia potencialmente reversibles antes de realizar un mielograma, como ser:
· endócrino-metabólicas: como la IRC o la patología tiroidea (frecuente asociación lesional en las EASs);
· farmacológica: suspender o disminuir empíricamente la dosis del fármaco sospechoso y revalorar;
· multicarencial: por déficit de B12, fólico, hierro.
Se debe realizar mielograma y eventual BMO una vez excluidas estas causas. La mayoría de los casos corresponden a anemias inflamatorias crónicas.
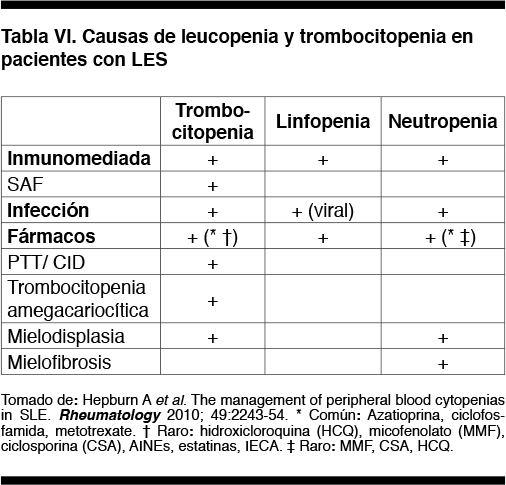
Características de la leucopenia y trombocitopenia asociadas al LES
La Tabla VI muestra las causas de trombopenia, linfopenia y neutropenia en pacientes con LES. La causa más frecuente es la inmunológica, periférica, manifestación de la propia enfermedad.(6) Estas citopenias son características del LES y siempre deben hacer sospechar esta entidad. La fluctuación en los recuentos celulares (neutropenias o plaquetopenias cíclicas), los rápidos cambios evolutivos (mejoría o peoría), una citopenia severa y de rápida instalación, orientan a la patogenia autoinmune. Sin embargo, es un diagnóstico de exclusión y se deben descartar las causas farmacológicas e infecciosas.(6)
Las citopenias autoinmunes pueden presentarse aisladas o asociadas. El síndrome de Evans consiste en la asociación de trombocitopenia y de anemia hemolítica y/o granulocitopenia autoinmunes.
Generalmente son citopenias leves y asintomáticas. La trombocitopenia es grave (< de 10.000 plaquetas) solo en 10% de los casos.(43) Se pueden producir ocasionalmente complicaciones severas como infecciones oportunistas, sepsis o sangrados.
Su presencia se correlaciona con actividad de la enfermedad y compromiso en otros órganos.(6,10) Por ejemplo, la linfopenia se asocia con insuficiencia renal/ glomerulonefritis y complicaciones del SNC en el LES.(10)
La trombocitopenia es un factor de riesgo independiente para el incremento de la mortalidad en el LES. (6, 44) Tienen implicancias terapéuticas ya que pueden cambiar el tratamiento global de la enfermedad según su severidad.(6)
Se han descrito leucopenia y linfopenia autoinmunes en la EMTC (Enfermedad Mixta del Tejido Conectivo) (20-75% y 76% respectivamente) y en el SSp (16% y 9% respectivamente)(37) Sin embargo, el hallazgo de leucopenia en pacientes con Esclerodermia debe hacer pensar en superposición con LES. La neutropenia puede preceder al diagnóstico de SSp (7%). Se ha descrito también linfopenia y neutropenia autoinmunes en AR (la neutropenia se asocia a esplenomegalia en el síndrome de Felty).
La trombocitopenia es infrecuente en el resto de las EASs a diferencia del LES.(37,45,46)
Las citopenias autoinmunes periféricas vinculadas a las EASs responden habitualmente al tratamiento con CE e IS. La hiperplasia medular es el hallazgo en la MO de estos pacientes, lo que sugiere destrucción periférica.(6)
No se hacen de rutina las determinaciones de anticuerpos antiplaquetarios, antineutrófilo y antilinfocito debido a su costo, disponibilidad limitada y demora en obtener los resultados.(6)
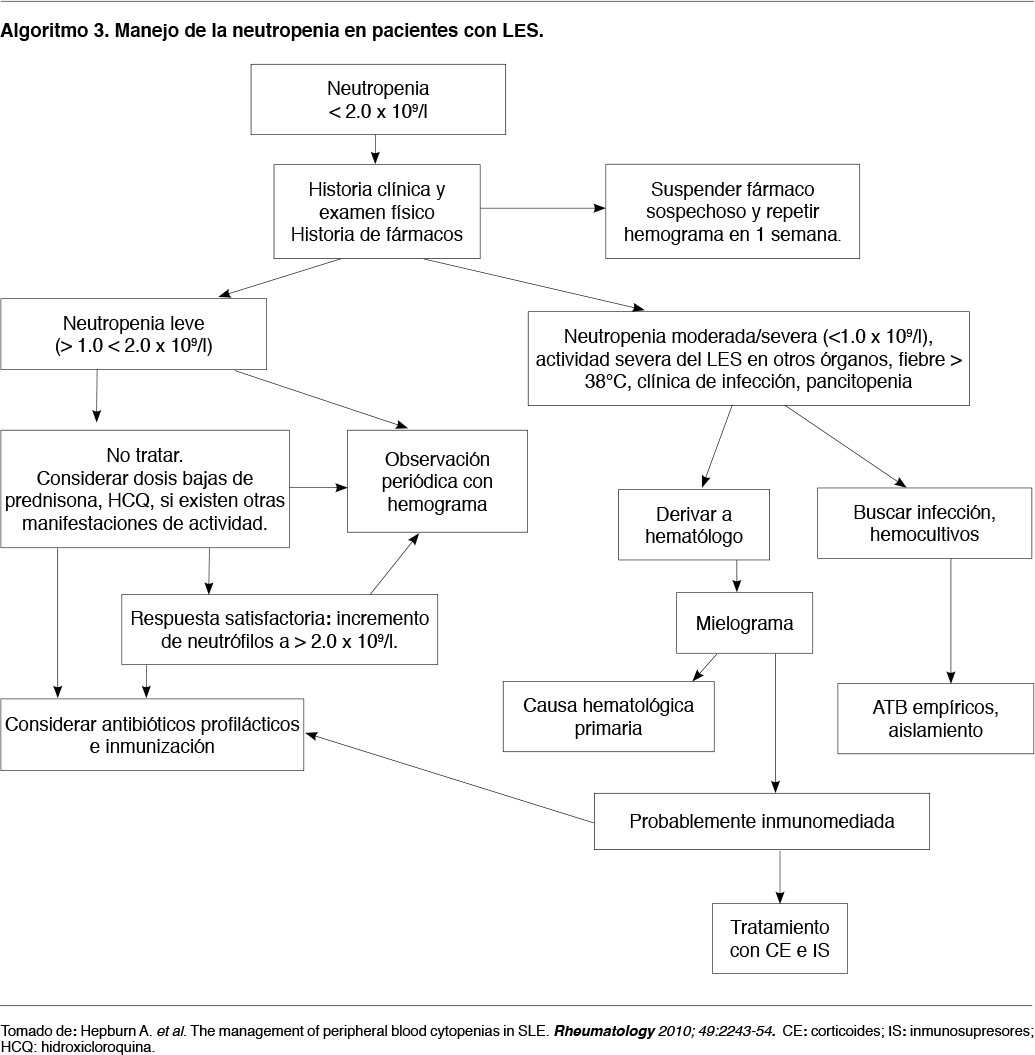
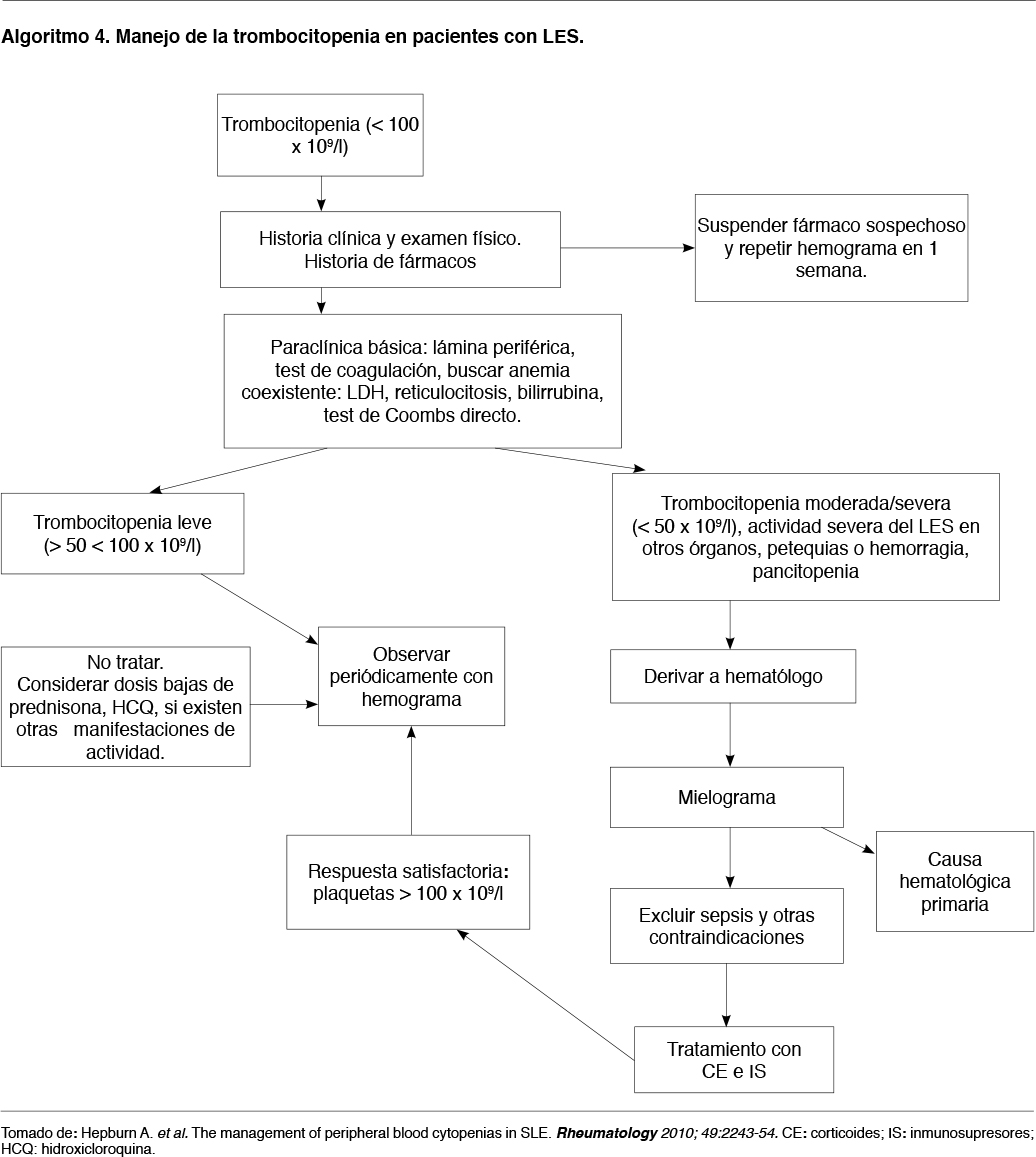
Los algoritmos 3 y 4 proponen una forma de manejo para la neutropenia y trombocitopenia respectivamente, en pacientes con LES.
Tratamiento de las citopenias de causa autoinmune en pacientes con LES
El tratamiento dependerá de la severidad del compromiso hematólogico y del balance lesional de la enfermedad.
Las citopenias son habitualmente leves en el LES y pueden no requerir tratamiento específico. El objetivo principal en estos casos es lograr la remisión de la enfermedad.(6,10) Sin embargo, el compromiso hematológico en algunos casos puede ser grave y condicionar la conducta terapéutica.
El tratamiento inicial en las citopenias severas de mecanismo autoinmunes son los corticoides (CE).
En la anemia hemolítica autoinmune se administra Prednisona oral entre 0,5 y 1 mg/kg peso/día durante 3 a 6 semanas. Habitualmente suele ser suficiente para controlar la citopenia clínicamente significativa, aunque no hay estudios controlados, randomizados, sobre su uso en citopenias en el LES. En caso de respuesta a CE, la dosis se reduce en forma progresiva en la evolución.
Si no existe respuesta o es parcial se pueden administrar pulsos de metilprednisolona intravenosos (250-1000 mg x 3 días).
Si la respuesta a los CE no es adecuada, o dependiendo de la severidad inicial de la citopenia o de una eventual cortico-dependencia (si se requieren más de 7,5 mg de Prednisona día para el control), se puede utilizar azatioprina a 2.5 mg/kg/día. Como fármacos de 2a línea pueden utilizarse micofenolato de mofetilo, micofenolato sódico, ciclosporina o metotrexate, no existiendo estudios randomizados y controlados sobre su uso.(6)
En casos con compromiso vital y sin respuesta a los pulsos de metilprednisolona (cortico-resistencia) se debe valorar la administración de inmunoglobulina intravenosa (400 mg/kg peso/día durante 3 días) y/o Rituximab.
En el caso de la trombocitopenia, debe descartarse en primer lugar la infección por H. pylori. Si el recuento plaquetario es superior a 50.000 plaquetas/mm3 sólo se realiza control evolutivo y no se indica tratamiento específico.
Si se encuentra entre 30.000 y 50.000/mm3 se indica Prednisona hasta 0,5 mg/kg peso/día, según gravedad clínica, con la intención de lograr cifras plaquetarias cercanas a 50.000/mm3. Si no hay respuesta se debe aumentar la dosis a 1 mg/kg peso/día.
Si las plaquetas son inferiores a 30.000/mm3 se indica Prednisona 1 mg/kg peso/día. Si no existe respuesta en 1 a 2 semanas se considera (en forma similar a la anemia hemolítica) indicar inmunoglobulina intravenosa o Rituximab.
En el caso de trombocitopenias inferiores a 10.000/mm3 o sangrado se indica Prednisona 2 mg/kg peso/día. Cuando existe compromiso vital se debe iniciar directamente con pulsos de metilprednisolona. Si no hay respuesta en 48 horas se administra inmunoglobulina intravenosa. Si estos tratamientos fracasan el siguiente paso sería la plasmaféresis y/o Rituximab.
La esplenectomía constituye el recurso terapéutico de necesidad en caso del fracaso de las medidas terapéuticas anteriormente señaladas tanto para la anemia hemolítica como para la trombocitopenia autoinmune.
En la anemia hemolítica microangiopática asociada al LES y después de descartar otras etiologías (hipertensión acelerada, fármacos, SAF catastrófico) las opciones terapéuticas son plasmaféresis, inmunoglobulina intravenosa y Rituximab.
El uso de inmunosupresores (IS) debe basarse en un adecuado balance riesgo beneficio, debido al riesgo de infección (sobre todo en neutropénicos severos) o de agravar las citopenias por su potencial efecto mielotóxico. El uso concomitante de G-CSF recombinante humano (rhG-CSF) y ATB puede reducir el riesgo de su uso.
El tratamiento de la anemia asociada a enfermedad crónica se basa en el control de la enfermedad de base. Algunos pacientes con anemia inflamatoria crónica y LES también pueden responder a la eritropoyetina humana recombinante (rHuEPO). Sin embargo en ocasiones se asocia a anticuerpos anti- rHuEPO que pueden inhibir tanto la rHuEPO administrada exógenamente como la EPO endógena causando APCR(5,47) y puede aumentar la respuesta inmunológica, en pacientes con LES. Por lo tanto, el tratamiento de esta anemia es el de la EASs, y el uso de EPO debería limitarse a pacientes con anemia sintomática y a los que requieren transfusión (situación infrecuente en la anemia inflamatoria).(5)
El uso del factor estimulante de colonias granulocíticas (rhG-CSF) en la neutropenia asociada al LES, debe restringirse a casos con infección severa y/o neutropenia grave (< 0,1 x 109/l) ya que se han observado empujes en AR y LES.(6,10,48) La mejoría puede ser rápida pero temporal por lo que se justifica su uso junto con IS. (6)
En pacientes neutropénicos febriles (< 1.0 x109/l) debe realizarse aislamiento y antibioticoterapia de amplio espectro.
Bibliografía
1. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
2. Petri M, Orbai A, Alarcón G, Gordon C, Merrill J, Fortin P, et al. Derivation and Validation of the Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus. Arthritis & Rheum 2012; 64(8): 2677-2686.
3. García A, Villegas A, González AF. Hematological abnormalities in patients with systemic lupus erythematosus. An Med Interna. 2002;19(10):539-43.
4. Beyan E, Beyan C, Turan M. Hematological presentation in SLE and its relationship with disease activity. Hematology. 2007;12(3):257-61.
5. S. Giannouli, Voulgarelis M, Ziakas P, Tzioufas A. Anaemia in SLE: from pathophysiology to clinical assessment. Ann Rheum Dis. 2006; 65(2):144-148.
6. Hepburn A, Narat S, Mason J. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology 2010; 2243-2254.
7. Mittal KK, Rossen RD, Sharp JT, Lidsky MD, Butler WT. Lymphocyte cytottoxic antibodies in SLE. Nature 1970;225:1255-6.
8. Butler WT, Sharp JT, Rossen Rd, Lidsky MD, Mittal KK, Gard DA. Relationship of the clinical course of systemic lupus erythematosus to the presence of circulating lymphocytotoxic antibodies. Arthritis Rheum 1972;15:251-8.
9. Winfield JB, Winchester RJ, Kunkel HG. Association of cold reactive antilymphocyte antibodies with lymphopenia in SLE. Arthritis Rheum 1975;18:587-94.
10. Nydegger U, García Carrasco M, Lior B, Orbach H, Newell L, Mankai A. Hematologic Autoimmune Diseases. En: Shoenfeld Y, Cervera R, Gershwin M, eds. Diagnostic Criteria in Autoimmune Diseases. Totowa, NJ: Humana Press, 2008:509-550.
11. Ruiz Arguelles A, Llorente L. The role of complement regulatory proteins (CD55 and CD59) in the pathogenesis on autoimmune hemocytopenias. Autoimmun Rev 2007;6:155-61.
12. Ronnblom L. Potential role of IFN α in adult lupus. Arthritis Res Ther 2010;12(Suppl. 1):S3.
13. Bengtsson AA, Sturfelt G, Truedsson L et al. Activation of type I interferon System in Systemic Lupus erythematosus correlates with disease activity but not with antirretroviral antibodies. Lupus 2000;9:664-71.
14. Nossent JC, Swaak AJ. Prevalence and significance of haematological abnormalities in patients with SLE. Q J Med 1991;80:605-12.
15. Hellmich B, Csernok E, de Hass M et al. Low Fcgamma receptor III and high granulocyte colony stimulating factor serum levels correlate with the risk of infection in neutropenia due to Felty´ s Syndrome or SLE. Am J Med 2002; 113:134-9.
16. Hellmich B, Csernok E, Schatz H, Gross WL, Schnabel A. Autoantibodies against granulocyte colony stimulating factor in Felty´s syndrome and neutropenic SLE. Arthritis Rheum 2002;46:2384-91.
17. Lipp E, Von Felten A, Sax H, Muller D, Berchtold P. Antibodies against platelet glycoproteins and antiphospholipid antibodies in autoimmune thrombocytopenia. Eur J Haematol 1998;60:283-8.
18. Michel M, Lee K, Piette JC et al. Platelet autoantibodies and lupus associated thrombocytopenia. Br J Haematol 2002;119:354-8.
19. Ziakas PD, Routsias JG, Giannouli S, Tasidou A, Tzioufas AG et al. Suspects in the tale of lupus associated thrombocytopenia. Clin Exp Immunol 2006;145:71-80.
20. Sultan SM, Begum S, Isenberg DA. Prevalence, patterns of disease and outcome in patients with SLE who develop severe haematological problems. Rheumatology 2003;42:230-4.
21. Fureder W, Firbas U, Nichol JL et al. Serum thrombopoietin levels and anti- thrombopoietin antibodies in SLE. Lupus 2002;11:221-6.
22. Faquin WC, Schneider TJ, Goldberg MA. Effect of inflammatory cytokines on hipoxia induced erythropoietin production. Blood 1992;79:1987-94.
23. Means RT, Krantz SB. Inhibition of human erythroid colony forming units by gamma interferon can be corrected by recombinant human erythropoietin. Blood 1991;78:2564-7.
24. Schooley JC, Kullgren B, Allison AC. Inhibition by interleukin 1 of the action of erythropoietin on erythroid precursors and its posible role in the pathogenesis of hypoplastic anaemias. Br J Haematol 1987;67:11-17.
25. Voulgarelis M, Kokori S, Ioannidis J, Tzioufas A, Kyriaki D, Moutsopoulos H et al. Anaemia in SLE: aetiological profile and the role of erythropoietin. Ann Rheum Dis 2000; 59:217-222.
26. Casadevall N, Dupuy E, Molho Sabatier P, Tobelem G, Varet B, Mayeux P. Autoantibodies against erythropoietin in a patient with pure red cell aplasia. N Engl J Med 1996. 334630-633. 633 [PubMed]
27. Casadevall N. Antibodies against rHuEPO: native and recombinant. Nephrol Dial Transplant 2002. 17(suppl 5) 42-47. [PubMed]
28. Chen Jl, Huang XM, Zeng XJ, Wang Y, Zhou MX, et al. Hematological abnormalities in SLE and clinical significance thereof: comparative analysis of 236 cases. Zhonghua Yi Xue Za Zhi. 2007. 22;87(19):1330-3.
29. Feng CS, Ng MH, Szeto RS, Li EK. Bone marrow findings in lupus patients with pancytopenia. Pathology 1991;23:5-7.
30. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J et al. Bone marrow findings in SLE patients with peripheral cytopenias. Clin Rheumatol 1998;17:219-22.
31. Oka Y, Kameoka J, Hirabayashi Y, Takahashi R, Ishii T, Sasaki T et al. Reversible bone marrow dysplasia in patients with SLE. Intern Med. 2008;47(8):737-42.
32. Actualización de la guía de práctica clínica para el manejo de la artritis reumatoide en España. Diciembre 2011. SER. Disponible en: http://www.ser.es/
33. Stein C. Fármacos inmunorreguladores. En: Ruddy S, Harris E, Sledge C, Budd R, Sergent J, eds. Kelley´s Textbook of Rheumatology, 6th ed. Philadelphia: WB Saunders, 2003: 879- 898.
34. Jiménez V, Gil V, Cervera R. Tratamiento de las Enfermedades Autoinmunes Sistémicas. Glucocorticoides e Inmunodepresores. Medicine 2005; 9(30):1977-1985.
35. Iwadate H, Kobayashi H, Yano K, Watanabe H, Ikeda K, Ogawa K et al. Therapy related myelodysplastic síndrome following cyclophosphamide pulse and/or methotrexate therapy in a patient with SLE. Fukushima J. Med. Sci. 2010;2: 121-127.
36. Fietta P, Delsante G, Quaini F. Hematologic manifestations of connective autoimmune diseases. Clin Exp Rheumatol. 2009; 27(1):140-54.
37. P. Manganelli, P. Fietta, F. Quaini. Hematologic manifestations of primary Sjögren’s syndrome. Clin Exp Rheumatol 2006; 24: 438-448.
38. Anderson LA, Pfeiffer RM, Landgren O, Gadalla S, Berndt SI, Engels EA. Risks of myeloid malignancies in patients with autoimmune conditions. Br J Cancer. 2009;100(5):822-8.
39. Baimpa E, Dahabreh IJ, Voulgarelis M, Moutsopoulos HM. Hematologic manifestations and predictors of lymphoma development in primary Sjogren Syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore). 2009;88(5):284-93.
40. Castro M, Conn DL, Su WP, Garton JP. Rheumatic manifestations in myelodysplastic Syndromes. J Rheumatol. 1991;18(5):721-7.
41. Nam EJ, Kang YM, Kang HR, Kim JH, Rho HJ, Lee MK et al. Rheumatoid Arthritis associated with Myelodysplastic Syndrome: a case report. J Korean Med Sci 1999;14:319-22.
42. Takashima H, Eguchi K, Origuchi T, Yamasita I, Nakashima M et al. Collagen diseases complicated with myelodysplastic síndrome (MDS)- reports of three cases. Ryumachi 1994;34(1):48-53.
43. Revenga M, Zea A. Lupus Eritematoso Sistémico. En: Carbonell J, Sociedad Española de Reumatología, eds. Semiología de las Enfermedades Reumáticas. Madrid: Editorial Médica Panamericana, 2006: 417- 428.
44. Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of SLE in a southern Chinese population. Rheumatology 2000;39:399-406.
45. Jiménez S, Aguiló S, Delgado G, Font J. Esclerosis Sistémica. Medicine 2005;9(30):1953-64.
46. Cervera R, Font J. Enfermedad Mixta del Tejido Conectivo. Medicine 2005;9(30):1974-76.
47. Bennett CL, Luminari S, Nissenson AR, Tallman MS, McWilliams N et al. Pure red cell aplasia and epoetin therapy. N Engl J Med 2004. 3511403-1408. 1408. [PubMed].
48. Vasiliu IM, Petri MA, Baer AN. Therapy with granulocyte colony stimulating factor in SLE may be associated with severe flares. J Rheumatol 2006;33:1878-80.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}