










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Medicina Interna
versión impresa ISSN 0250-3816versión On-line ISSN 1688-423X
Arch. Med Int vol.36 no. 2 Montevideo jul. 2014
Caso clínico de interés
Sarcoidosis aguda
Variante de Síndrome de Löfgren sin eritema nodoso
Acute sarcoidosis
Erythema Nodosum-Free Variant of Löfgren’s Syndrome
Dr. Gerardo Pérez,
Ex Asistente de Clínica Médica “C”. Facultad de Medicina. UdelaR. Departamento Clínico de Medicina, Hospital Británico. Montevideo.
Dr. Jorge Facal
Profesor Titular Clínica Médica “1”. Hospital Maciel. Facultad de Medicina. UdelaR. Departamento Clínico de Medicina, Hospital Británico. Montevideo.
Recibido: 05/11/03 - Aceptado: 14/03/14
Centro de Trabajo: Departamento Clínico de Medicina, Hospital Británico. Montevideo.
Correspondencia: Dr. Gerardo Pérez, Hospital Británico. Av. Italia 2420. Montevideo, Uruguay. Email: gerper4@gmail.com
RESUMEN: Arch Med Interna 2014 - 36(2):79-83
El síndrome de Löfgren, es una variante aguda de la sarcoidosis, que se caracteriza por fiebre, eritema nodoso, adenomegalias hiliares pulmonares y artritis. En general, tiene un curso benigno y autolimitado, que contrasta con las formas crónicas que requieren uso de corticoides y tienen tendencia a la recidiva. Se describe aquí el caso clínico de un paciente joven, de sexo masculino, con artritis pero sin eritema nodoso, lo que dificultó el planteo diagnóstico de síndrome de Löfgren. Se realiza además una breve descripción comparativa entre la presentación clínica de la sarcoidosis crónica y el síndrome de Löfgren.
Palabras clave: artritis, eritema nodoso, sarcoidosis, Löfgren
ABSTRACT: Arch Med Interna 2014 - 36(2):79-83
Löfgren syndrome is an acute variant of sarcoidosis, and is defined by erythema nodosum, hilar lymphadenopathy and arthritis. Usually shows a benign self-limited course contrasting with chronic sarcoidosis with persistent relapsing presentations requiring corticosteroids most of the times. This article describes a young male patient with Löfgren syndrome with the distinct fact of the absence of erythema nodosum, which initially made sarcoidotic etiology of arthritis more difficult to think of. A brief comparative description of sarcoidosis and Löfrgen syndrome features is also made.
Keywords: arthritis, erythema nodosum, sarcoidosis, Löfgren
Introducción
La historia moderna de la sarcoidosis se remonta a 1899, cuando el dermatólogo Noruego Caesar Boeck acuña el término para describir nódulos cutáneos caracterizados por focos compactos y definidos de “células epitelioides con grandes núcleos pálidos y también algunas células gigantes”. Teniendo en mente su semejanza con los sarcomas, Boeck denominó a esta condición, “sarcoma benigno múltiple de la piel”(1).
La sarcoidosis es una enfermedad multisistémica de etiología desconocida que puede afectar cualquier órgano o sistema en el cuerpo, y se caracteriza por la presencia de granulomas no caseificantes en las muestras biopsicas(2).
Caso clínico
Paciente de sexo masculino, 38 años, ex fumador, trabajador rural (trabaja con ganado bovino). Sin otros antecedentes a destacar.
Consulta por fiebre y artralgias de 15 días de evolución. Artralgias en tibiotarsianas bilateral con signos fluxivos, agregando posteriormente artritis radiocarpiana bilateral. Fiebre hasta 38,5ºC, con chuchos de frío. Deposiciones liquidas 48 horas antes del inicio de las artralgias. Al momento de la consulta el paciente no presentaba ninguna manifestación digestiva o respiratoria. Tampoco presentaba síntomas sugestivos de uretritis o uveítis. Al examen físico, destacaba un paciente con buen estado general, febril (38ºC), eupneico, PA 130/70 mmHg. Del examen físico a nivel de piel y mucosas no presentaba anemia clínica, ni lesiones hemorragiparas. Rubor, calor, tumefacción e intenso dolor con maniobras de movilización tanto activas como pasivas a nivel tibiotarsiano bilateral y en menor grado, en ambas articulaciones radiocarpianas. No lesiones compatibles con eritema nodoso. La exploración cardiovascular, pleuropulmonar y abdominal no tenían elementos patológicos a destacar.
De la paraclínica se señala el hemograma sin alteraciones a nivel de las 3 series, proteína C reactiva elevada con valor de 48,5 mg/L, VES también elevada de 46 mm/1ª hora, función renal y hepatograma normales, proteinograma electroforético normal, calcemia y calciuria normales.
Los hemocultivos y urocultivos no presentaron desarrollo, el exudado faríngeo desarrolló flora normal, mientras que la determinaión de antiestreptolisina O (AELO) fue menor a 200 UI/ml (no reactivo). El coprocultivo descartó la presencia de Salmonella o Shigella. Las serologías para hepatitis B, C y VIH fueron no reactivas, al igual que el estudio para lues. Por el antecedente ocupacional con contacto con ganado se solicitó serología para brucelosis, con resultado negativo. Las serologías para Chlamydias también fueron negativas.
La complementemia fue normal, mientras que los anticuerpos antinucleares (ANA), anticitoplasma de neutrófilo (ANCA), factor reumatoideo y anticuerpos antipéptidos citrulinados también fueron negativos.
La dosificación de enzima conversora de angiotensina (ECA) se encontró en rango normal: 35 U/L (VN: 8-55).
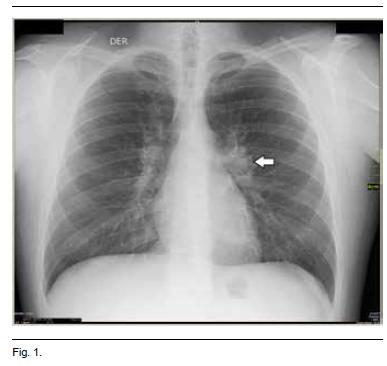
A nivel imagenológico se solicitaron radiografías de articulaciones tibiotarsianas y puños que fueron normales. La radiografía de tórax evidencio agrandamiento de los hilios compatible con adenomegalias (Figura 1).
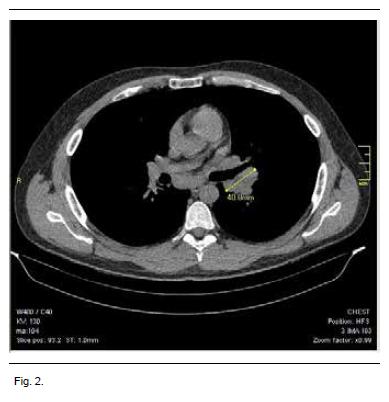
Se complementó con tomografía de tórax que evidencia adenopatías hiliares bilaterales y paratraqueales altas y bajas a derecha, que no forman conglomerados (Figura 2).
Con planteo clínico-radiológico de sarcoidosis de presentación aguda a forma de síndrome de Löfgren se inició tratamiento con prednisona a dosis de 40 mg al día. A las 36 horas el paciente se encontraba subjetivamente mejor, sin fiebre y con marcada disminución del componente fluxivo en las articulaciones comprometidas. Se otorgó el alta hospitalaria con prednisona en estas dosis durante 2 meses con posterior descenso gradual de ella.
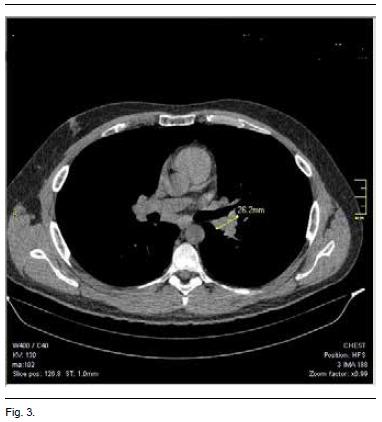
A los 2 meses del alta el paciente se encuentra asintomático. Se realizó una TAC de tórax de control, que muestra una disminución importante del tamaño de las adenopatías (Figura 3). A la fecha, el paciente no ha mostrado signos de recidiva, y se inició el descenso de la dosis de corticoides.
características Comparativas entre sarcoidosis y del síndrome de Löfgren
Sarcoidosis crónica
Epidemiología
La sarcoidosis puede afectar a personas de cualquier raza o grupo étnico y de cualquier edad, aunque en general se manifiesta antes de los 50 años con un pico máximo entre los 20 y 39 años(3). La prevalencia de la enfermedad se estima en 1 a 40 casos cada 100.000 habitantes, con una tasa de incidencia anual de 10.9 cada 100.000 para la raza blanca y 35,5 cada 100.000 para afrodescendientes(2). En este último grupo racial, la sarcoidosis tiene mayor riesgo de cronicidad y de un curso fatal(4). Si bien el nivel socioeconómico no es un factor de riesgo para el desarrollo de sarcoidosis, se observó que niveles socioeconómicos bajos se corresponden con formas más agresivas de la enfermedad al momento de su presentación. Esta diferencia por nivel sociocultural se observa aun luego de ajustar las diferencias por raza, sexo y edad(5).
Patogenia
Se plantea como posible causa desencadenante la exposición a antígenos aéreos en función de que la enfermedad frecuentemente afecta aparato respiratorio, ojos y piel. Numerosos estudios establecen asociación entre exposición a antígenos aéreos tanto orgánicos(6) como inorgánicos(7). Recientemente, se publicó un trabajo que demuestra la mayor incidencia de sarcoidosis entre los bomberos y rescatistas del atentado al World Trade Center en setiembre de 2001(8). Estos potenciales antígenos actuarían en individuos predispuestos por susceptibilidad genética. El primer reporte relacionando sarcoidosis y marcadores genéticos fue la asociación entre la presencia de antígenos clase I HLA-B8 y desarrollo de sarcoidosis aguda(9). Como la potencialidad de manifestar sarcoidosis depende de la susceptibilidad genética y su interacción con los modificadores (antígenos ambientales), se busca una vinculación entre ambos para intentar definir la causa (o causas) de la enfermedad. A la fecha, se estableció la interacción entre el locus de susceptibilidad de sarcoidosis HLA DQB1 y ambientes con altos niveles de humedad(10).
Manifestaciones clínicas
La sarcoidosis puede presentarse con manifestaciones casi en cualquier órgano o sistema, pero los pulmones son los órganos más frecuentemente afectados (80%)(2). En más del 90% de los casos, la sarcoidosis se manifiesta por adenomegalias intratorácicas, compromiso pulmonar, oftalmológico o cutáneo, o combinación de estas lesiones. Otras manifestaciones frecuentes incluyen uveítis, hepatoesplenomegalia y polidenomegalias. El diagnóstico surge frecuentemente a partir del hallazgo de anormalidades en la radiografía de tórax de rutina. Es frecuente la presencia de síntomas sistémicos como astenia, sudoración nocturna y pérdida de peso(1). El diagnóstico de sarcoidosis requiere la identificación de granulomas no caseificantes en muestras biópsicas, junto a un cuadro clínico compatible, y la exclusión de otras causas de enfermedad granulomatosa(11).
El tratamiento se basa en corticoides (sistémicos, inhalatorios o tópicos) y/o inmunomoduladores (metotrexate, hidroxicloroquina, azatioprina, entre otros). La mayoría de los pacientes no los requiere y la indicación depende fundamentalmente de la existencia de riesgo de deterioro funcional del órgano u órganos afectados.
El pronóstico del paciente con sarcoidosis depende de la etapa de la enfermedad al momento de la presentación, definido por el grado de afectación pulmonar (infiltrados) o la presencia de ganglios hiliares pulmonares.
En cuanto al pronóstico, dos tercios de los pacientes logran la remisión dentro de la primera década del diagnóstico, con poca o ninguna consecuencia funcional en los órganos afectados. Aproximadamente el 50% de estos pacientes logran la remisión dentro de los 3 primeros años luego del diagnóstico. Desafortunadamente un tercio de los pacientes presentan una forma de sarcoidosis de curso implacable con severo compromiso orgánico. La muerte por sarcoidosis se produce en menos del 5% de los pacientes, como consecuencia de fibrosis pulmonar con insuficiencia respiratoria severa. Otras causas de muerte son el compromiso neurológico y cardíaco(1).
Síndrome de Löfgren
La primera descripción del síndrome se atribuye a Sven Löfgen en 1953 con una serie de 113 pacientes, donde además se reconoce por primera vez este síndrome como una variante aguda de sarcoidosis. A partir de ese momento se define por la presencia de adenomegalias hiliares pulmonares bilaterales, eritema nodoso y artralgias (frecuentemente asociado a artritis) principalmente de grandes articulaciones. La sarcoidosis se presenta como síndrome de Löfgren en porcentajes que oscilan entre 9 y 35%(12,13). Epidemiológicamente, es más frecuente en determinadas áreas geográficas y razas. Las mujeres jóvenes de procedencia escandinava tienen la mayor incidencia, mientras que los afrodescendientes tienen la menor incidencia de síndrome de Löfgren(14).
Patogénicamente, el síndrome de Löfgren presenta una fuerte asociación con la presencia del marcador genético HLA-DRB1*03. La positividad o negatividad de este marcador tiene implicancias en el curso evolutivo de la enfermedad(15).
La presentación típica del síndrome, como fue descrito, incluye la asociación de adenopatías hiliares, poliartritis aguda y eritema nodoso, habitualmente en el contexto de fiebre. En una serie de 186 pacientes con síndrome de Löfgren, la frecuencia de síntomas y signos fueron: artralgias (68%), fiebre (38%), disnea (13%), eritema nodoso (13%), hepatomegalia (6%)(14).
La oligoartritis aguda que acompaña al síndrome de Löfgren usualmente afecta a las articulaciones tibioperonea y tibiotarsiana en forma bilateral (> 90% de los casos). Se pueden afectar otras grandes articulaciones de miembros inferiores y muchas veces lleva a confusión diagnóstica con una artritis reactiva(15). En un estudio prospectivo de pacientes con artritis de reciente inicio se encontró que la presencia de artritis con menos de 2 meses de evolución, con afectación simétrica de tobillos, en pacientes menores de 40 años tiene una alta sensibilidad (85%) y especificidad (99%) para el diagnóstico de artritis por sarcoidosis(16).
En general, la artritis del síndrome de Löfgren es autolimitada, pero aproximadamente 30% de los pacientes muestran una evolución persistente con síntomas que sobrepasan los 2 años de evolución(17). A pesar de esto, la artritis raramente reaparece una vez resuelto el episodio inicial(18).
La oligoartritis en el paciente del caso analizado se comportó típicamente como la artritis del síndrome de Löfgren con un compromiso predominante a nivel tibiotarsiano bilateral. El antecedente de deposiciones líquidas previo al inicio de la artritis hizo plantear una artritis reactiva (síndrome de Reiter). El escaso lapso temporal entre los síntomas digestivos y la artritis (aprox. 48 horas) alejaron esta posibilidad, ya que en el Reiter las manifestaciones digestivas suelen preceder en 1 a 3 semanas el inicio de las artralgias, así como también la ausencia de lesiones cutáneas palmo-plantares típicas del mismo (queratoderma blenorrágica).
Clásicamente, se considera la aparición de eritema nodoso en este contexto como el elemento fundamental para el planteo diagnóstico de síndrome de Löfgren. Ha sido debatido si los pacientes deben necesariamente presentar eritema nodoso para ser considerados como portadores de síndrome de Löfgren o si existe una variante del síndrome sin eritema nodoso.
El eritema nodoso puede responder en realidad a múltiples etiologías. En una serie de García-Porrua se encontró que de 106 pacientes con eritema nodoso confirmado por biopsia, 34% fueron formas idiopáticas, 22% correspondieron a sarcoidosis o síndrome de Löfgren, 20% secundario a infecciones virales del tracto respiratorio, 7% secundario a infección por estreptococo beta hemolítico grupo A (EBHA), 5% secundario a tuberculosis, 3% secundarios a drogas (anticonceptivos) y correspondiendo el resto a enfermedades autoinmunes y neoplasias(19).
En un estudio de 150 pacientes con presentación clínica de síndrome de Löfgren, se observó que la presencia de eritema nodoso es más frecuente y constante en las mujeres en comparación con los hombres (67% vs 27%; p < 0.0001)(15). No se encontró diferencia en el resto de las manifestaciones clínicas según el sexo. Se concluye que las manifestaciones del síndrome de Löfgren difieren entre hombres y mujeres, con presencia de eritema nodoso predominantemente en mujeres y marcada artritis de tobillos sin eritema nodoso en hombres(1,15,20).
La ausencia de eritema nodoso en el caso analizado, que inicialmente podría alejar el planteo de sarcoidosis aguda, en realidad no lo invalidad, ya que la presentación fue la más típica del Löfgren en el sexo masculino con franco predominio de artritis asociado a adenopatías hiliares y sin eritema nodoso.
Las linfadenopatias torácicas del síndrome de Löfgren típicamente se presentan a nivel hiliar bilateral y paratraqueal derecho. Muestran tendencia a resolverse en plazo de semanas en el 90% de los casos, con solo pocos casos descritos con persistencia y progresión(18).
El control evolutivo del paciente a los 2 meses mostró una franca disminución del tamaño de las adenopatías, acorde con la forma evolutiva de esta variante de sarcoidosis.
En definitiva, si bien el eritema nodoso es el hallazgo que nos conduce habitualmente a la sospecha de sarcoidosis/Löfgren, su ausencia en un paciente de sexo masculino que cursa una oligoartritis de miembros inferiores febril, debe hacer sospechar la posibilidad del diagnóstico mencionado.
Diagnóstico de Síndrome de Löfgren
El diagnóstico de síndrome de Löfgren depende de la presencia de al menos 2 de 3 de los hechos clínicos que lo definen (eritema nodoso, adenopatías hiliares bilaterales y oligoartritis).
Se discute la indicación de biopsia para obtener confirmación histológica. La sarcoidosis, cualquiera sea su variante (incluído el síndrome de Löfgren), no es la única enfermedad que cursa con adenopatías hiliares. Los diagnósticos diferenciales incluyen infecciones fúngicas (histoplasmosis, coccidioidomicosis), linfomas, carcinoma broncogénico y tuberculosis(21). Por esta razón, algunos autores plantean que el diagnóstico de sarcoidosis se sustenta en un cuadro clínicor-radiológico compatible y la presencia de granulomas no caseificantes en uno o más órganos, con ausencia de microorganismos o partículas(1). Las principales sociedades científicas en enfermedades respiratorias también recomiendan que el diagnóstico de sarcoidosis se confirme histológicamente(22).
Sin embargo, existe acuerdo en que la presentación clínica de sarcoidosis aguda a forma de síndrome de Löfgren no requiere confirmación histológica siempre que la evolución sea rápida y favorable(1, 22). La presentación mencionada con adenopatías hiliares y otros hallazgos típicos de sarcoidosis tiene un valor predictivo positivo de 99% y por tanto, suficiente para brindar una aceptable certeza diagnóstica. La realización de una biopsia en este contexto resulta innecesaria(23).
Desde el punto de vista del laboratorio, no existe prueba que confirme el diagnóstico de sarcoidosis en cualquiera de sus variantes. Varios autores mencionan la utilidad de los niveles plasmáticos de ECA en el diagnóstico de sarcoidosis. La ECA se eleva en la sarcoidosis en aproximadamente 50% de los casos(2), pero además de ser poco sensible, es también inespecífica, ya que puede encontrarse elevada también en otros procesos inflamatorios(1,22). Tampoco resulta útil como parámetro de actividad de la enfermedad y/o respuesta terapéutica(1).
Tratamiento del síndrome de Löfgren
Debido a que la artritis aguda en esta etiología es habitualmente autolimitada, la mayoría de los autores concuerdan que el tratamiento inicial sea en base a antiinflamatorios no esteroideos (AINEs) en monoterapia. Si luego de dos semanas de uso son inefectivos o mal tolerados, entonces estarían indicados otros agentes.
En general, los glucocorticoides y dentro de estos, la prednisona, es el agente de elección en dosis de 20 a 40 mg al día(1). Estas dosis se mantienen por 1 a 3 meses para luego iniciar un descenso progresivo si la situación clínica del paciente lo permite. En casos excepcionales, se requiere el uso de otros agentes como hidroxicloroquina o metotrexate.
El pronóstico del paciente con síndrome de Löfgren es excelente, incluso en aquellos sin tratamiento. A 2 años del diagnóstico, solo 8% de los pacientes tienen evidencia de actividad de la enfermedad y solo 6% presenta recaídas (porcentaje mucho menor que en la sarcoidosis crónica)(14).
Bibliografía
1. Iannuzzi M, Rybicki N, Teirstein A. Sarcoidosis. N Engl J Med 2007; 357:2153-65.
2. Belfer MH, Stevens RW. Sarcoidosis: a primary care review. Am Physician 1998; 58:2041-50.
3. Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol 1997; 145:234-41.
4. Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001 ;164: 1885-9.
5. Rabin DL, Thompson B, Brown KM, et al. Sarcoidosis: social predictors of severity at presentation. Eur Respir J 2004; 24:601-8.
6. Bresnitz EA, Strom BL. Epidemiology of sarcoidosis. Epidemiol Rev 1983; 5: 124-56.
7. Newman LS, Rose CS, Bresnitz EA, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med 2004; 170: 1324-30.
8. Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest 2007; 131:1414-23.
9. Brewerton DA, Cockburn C, James DC, James DG, Neville E. HLA antigens in sarcoidosis. Clin Exp Immunol 1977;27: 227-9.
10. Iannuzzi MC, Maliarik MJ, Poisson LM, Rybicki BA. Sarcoidosis susceptibility and resistance HLA-DQB1 alleles in African Americans. Am J Respir Crit Care Med 2003; 167:1225-31.
11. Johns CJ, Michele TM. The Clinical management of sarcoidosis. A 50-year experience at the Johns Hopkins Hospital. Medicine (Baltimore). 1999; 78: 65-111.
12. Siltzbach LE, James DG, Neville E, et al. Course and prognosis of sarcoidosis around the world. Am J Med 1974;57:847-52.
13. Moore AL. Löfgren´s syndrome and arthritis. J Fam Pract 1981; 12: 1071-2.
14. Mana J, Gomez-Vaquero C, Montero A. Lofgren´s syndrome revisited: a study of 186 patients. Am J Med. 1999; 107: 240-5.
15. Grunewald J, Eklund A. Löfgren’s Syndrome Human Leukocyte Antigen Strongly Influences the Disease Course. Am J Respir Crit Care Med 2009 (179): 307–312.
16. Visser H, Vos K, Zanelli E. Sarcoid arthritis: clinical characteristics, diagnose aspects and risk factors. Ann Rheum Dis 2002; 61:499.
17. Johard U, Eklund . Recurrent Löfgren´s syndrome in three pateints with sarcoidosis. Sarcoidosis 1993; 10:25
18. Sequeira W, Aggarwal R. Sarcoid arthropathy. UpToDate 2013. En: http://www.uptodate.com/contents/sarcoid-arthropathy. Acceso: 28/5/2013.
19. Garcia-Porrua C, Gonzalez-Gay MA, Vazquez-Caruncho M. Erythema nodosum: etiologic and predictive factors in a defined population. Arthritis Rheum 2000; 43: 584-92.
20. Idali F, Wikén M, Wahlström J, et al. Reduced Th1 response in the lungs of HLA-DRB1*0301 patients with pulmonary sarcoidosis. Eur Respir J 2006;27:451-9.
21. Goroll AH, Mulley AG.: Office evaluation and management of the adult patient. Primary care medicine 4th ed. Philadelphia: Lippincot Williams & Wilkins; 2000.
22. American Thoracic Society (ATS), European Respiratory Society (ERS) and World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG). Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999; (160): 736–755.
23. Reich JM, Brouns MC, O´Connor EA, Edwards MJ. Mediastinoscopy in patients with presumptive stage I sarcoidosis: a risk/benefit, cost/benefit analysis. Chest 1998; 113: 147-53.

