










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Medicina Interna
Print version ISSN 0250-3816On-line version ISSN 1688-423X
Arch. Med Int vol.35 no.3 Montevideo Dec. 2013
Caso clínico de interés
Descripción de un caso de fiebre mediterránea familiar
Family Mediterranean Fever - case description Dra. Fabiana De Armas
Residente de Medicina Interna Facultad de Medicina. UdelaR. CASMU. Montevideo.
Dr. Álvaro Díaz Berenguer
Profesor Agregado de Clínica Médica. Facultad de Medicina. UdelaR. CASMU. Montevideo.
Dr. Víctor Raggio
Profesor Adjunto del Departamento de Genética, Facultad de Medicina. UdelaR. Montevideo.
Dr. Carlos Dufrechou
Profesor Director de Clínica Médica “2”. Facultad de Medicina. UdelaR. Montevideo.
Dra. Beatriz Goja
Jefa del Departamento de Medicina Interna de CASMU. Montevideo.
Recibido: 04/04/13-Aceptado: 27/10/13
Correspondencia: Dra. Fabiana de Armas Mutuberría. Tomás Gomensoro 2907, CP11600. Tel. 2487 0754; Cel. 099590144 e-mail: dearmasf@adinet.com.uy
RESUMEN: Arch Med Interna 2013 - 35(3):101-104
La fiebre mediterránea familiar es una enfermedad autosómica recesiva, causada por mutaciones en el gen MEFV, el cual codifica una proteína denominada pirina. Esta proteína interviene en la formación del inflamasoma que estimula la activación de la IL-1β a través de la activación de la caspasa-1. Esta proteína tendría un efecto inhibitorio sobre la activación de la IL-1 β, por lo que las mutaciones de pérdida de función de ésta causarían una regulación en más de la inflamación. Forma parte de los síndromes hereditarios de fiebre periódica, caracterizados por episodios inflamatorios recurrentes autolimitados que cursan con fiebre, poliserositis, sinovitis y manifestaciones cutáneas. Es más frecuente en determinados grupos étnicos como es el caso que se presenta en este trabajo, se considera de interés por la dificultad en su diagnóstico. Es opinión de los autores que esta enfermedad es subdiagnosticada en Uruguay.
Palabras clave: Fiebre mediterránea familiar, enfermedades autoinflamatorias hereditarias.
ABSTRACT: Arch Med Interna 2013 - 35(3):101-104
Familial Mediterranean fever is an autosomal recessive disease caused by mutations in the gene MEFV, which encodes a protein called pirin. This protein is involved in the formation of stimulating inflamasome activation of IL-1β through activation of caspase-1. This protein would have an inhibitory effect on activation of IL-1β, so loss of function mutations cause an up-regulation of the inflammatory response. It is part of the hereditary periodic fever syndromes which characterized by recurrent episodes of fever, polyserositis, synovitis, and cutaneous manifestations.
It is more common in certain ethnic groups such as this case, which is presented because of the difficulty in its diagnosis. It is the opinion of the authors that this disease is underdiagnosed in Uruguay.
Keywords: familial mediterranean fever, hereditary autoinflammatory diseases.
INTRODUCCIÓN
La fiebre mediterránea familiar (FMF) pertenece al grupo de las fiebres periódicas hereditarias, que se caracterizan por presentar episodios febriles recurrentes y autolimitados con poliserositis, sinovitis y manifestaciones cutáneas. Fue descrita en 1908 por Janeway y Mosenthal (1) y en 1955 Heller (2) introdujo por primera vez el término de fiebre mediterránea familiar debido a la presentación familiar y a su distribución geográfica (3).
Las fiebres periódicas son entidades autoinflamatorias (4), un grupo de enfermedades no infecciosas, sin sustrato autoinmune, alérgico, ni asociado a inmunodeficiencias.
Es una enfermedad de herencia autosómica recesiva y afecta principalmente a descendientes de poblaciones mediterráneas, como los judíos sefardíes, asquenazíes, armenios, árabes y turcos. La frecuencia de portadores se ha calculado de 1:5 a 1:7 y la frecuencia de enfermos llegaría a 1/200 individuos en algunas de estas poblaciones. Se estima que hay 100.000 afectados en todo el mundo (5) y se presenta en el 90% de los casos antes de los 20 años de edad (6), siendo más frecuente en hombres. El número de armenios en Uruguay asciende a 19.000, por lo que sólo en esa población habría aproximadamente 100 casos (7).
Clínicamente, se manifiesta por episodios recurrentes de fiebre que puede alcanzar 40ºC, acompañados de otros síntomas inflamatorios a forma de dolor abdominal agudo, que muchas veces es difícil de diferenciar de cuadros quirúrgicos. La peritonitis puede ser periódica y formar parte de un síndrome de poliserositis con compromiso de pleura, pericardio y articulaciones. Otra manifestación característica es la formación de un eritema erisipeloide en cara anterior de pierna y dorso de pie (8). Se pueden asociar mialgias, signos de escroto agudo por inflamación de la túnica albugínea y meningitis aséptica (9,10). Se ha encontrado asociación con vasculitis de Shöinlein-Henoch, poliarteritis nodosa y enfermedad de Behçet (11). Entre los episodios febriles los pacientes cursan asintomáticos y recurren en forma irregular. Los episodios febriles ocurren sin desencadenantes evidentes, aunque algunos pacientes lo relacionan con estímulos físicos, la exposición al frío, el estrés emocional o el ciclo menstrual. Los exámenes de laboratorio durante los ataques revelan elevación de los reactantes de fase aguda. La FMF está producida por mutaciones en el gen MEFV situado en el locus 16p13, mapeado en 1997 (12,13). Se han identificado hasta la fecha más de 150 mutaciones del gen (14). Las mutaciones más frecuentes son M694V, V726A (exón 10) y el E148Q (exón 2). La mutación M694V muestra una asociación significativa con el desarrollo de amiloidosis renal al igual que la mutación compuesta V726A-E148Q, por lo que es un determinante de peor pronóstico (15).
Este gen codifica una proteína de 781 aminoácidos, denominada Pirina (“fiebre y fuego” en griego) por el Internacional FMF Consortium (16) y Marenostrina (“mare nostrum”) por el French FMF Consortium(17). Dicha proteína es expresada en el citoplasma de monocitos maduros en asociación con los microtúbulos, pero se encuentra fundamentalmente en el núcleo de granulocitos, células dendríticas y fibroblastos sinoviales (18). Además la pirina participa en la vía del factor nuclear kB (NFkB), regulador central de genes de inflamación y apoptosis (19). Esta proteína tendría un efecto inhibitorio sobre la activación de la IL-1β, por lo que las mutaciones de pérdida de función de ésta causarían una regulación en más de la inflamación. El pronóstico de la enfermedad está íntimamente relacionado con la aparición de amiloidosis renal, secundaria a los repetidos procesos de inflamación, que constituye la complicación más grave y se relaciona con el origen étnico (judíos sefardíes), el genotipo y el tratamiento inadecuado (20).
HISTORIA CLÍNICA
Paciente de 75 años de edad, hijo y nieto de armenios por vía paterna y materna. Antecedentes patológicos de hipertensión arterial y tabaquismo, lesiones hepáticas conocidas desde hace 30 años e interpretadas como quistes hidáticos calcificados.
Presenta historia de artralgias de miembros inferiores desde la niñez, con episodios febriles con temperatura axilar de hasta 40ºC, de 1 a 3 días de duración, que cedían espontáneamente, ocasionalmente acompañados de dolor en hipocondrio derecho. Los episodios se repetían en forma periódica cada 2 semanas a 2 meses. Entre dichos episodios el paciente se encontraba asintomático. Refiere que su hermano menor presentaba sintomatología similar pero más leve.
Ingresó en 1996 en el Departamento de Medicina Interna por fiebre de 20-30 días de evolución, mialgias y astenia. En esa oportunidad con planteo de fiebre de origen desconocido, se realizaron los siguientes exámenes paraclínicos: velocidad de eritrosedimentación 140 mm 1ª hora, hemocultivos, urocultivo y coprocultivo sin desarrollo bacteriano. Baciloscopías y PPD negativos. Serología para lúes, Micoplasma pneumoniae, Chlamydias, fiebre Q, citomegalovirus, toxoplasma y brucelosis no reactivos. Coproparasitario negativo. Anticuerpos antinucleares, factor reumatoideo, complejos inmunes circulantes y Coombs directo negativos. Crioaglutininas a 4ºC positivo 1/8, a 22ºC y 27ºC negativos. Proteinograma electroforético y enzimograma muscular normales. Fondo de ojo normal. Radiografía de tórax y ecocardiograma transesofágico normales. Tomografía computada (TC) de abdomen: calcificaciones hepáticas y esplénicas; fibrolaparoscopía con biopsia hepática mostró esteatosis y fibrocolonoscopía no presentó anormalidades. Ecodoppler venoso de miembros inferiores descartó trombosis. Se realizó consulta con odontólogo quien extrajo resto dentario.
En la evolución presentó un eritema erisipeloide en cara anterior de pierna, la biopsia de piel informó escaso infiltrado linfoide en dermis y la de músculo esquelético fue normal.
La fiebre y los síntomas remitieron espontáneamente, otorgándose el alta, sin diagnóstico etiológico.
Valorado en forma ambulatoria en 1999 por dolor abdominal recurrente con ecografía abdominal que mostró imágenes hepáticas ya conocidas y un pólipo vesicular. La paraclínica humoral de ese momento fue normal. Se realizó colecistectomía de coordinación. La anatomía patológica de la pieza extraída informó: colecistopatía crónica moderadamente fibroproductiva con adenomiosis parietal asociada, de moderada entidad.
El paciente ingresó nuevamente en 2003 por cuadro agudo de abdomen sin alteraciones humorales; en TC de abdomen: engrosamiento circunferencial de las paredes del colon ascendente de aspecto tumoral, con algunos tractos densos en la grasa adyacente con sospecha de infiltración. Hígado con pequeñas calcificaciones ya conocidas que pueden corresponder a granulomas. Bazo con algunos granulomas calcificados. Resto del estudio sin alteraciones. En la fibrocolonoscopía se observaron hemorroides internas y papilas hipertrofiadas. Con estos hallazgos se sometió al paciente a una laparotomía exploradora cuyo diagnóstico postoperatorio fue de peritonitis plástica adherencial, de etiología desconocida. Biopsia epiplón y peritoneo informó: tejido fibroadiposo, vascularizado con vasos congestivos, focos de hemorragia y mínimo infiltrado linfocitario; con revestimiento discontinuo por células mesoteliales aplanadas. Ausencia de elementos de malignidad en el material estudiado. Con buena evolución y asintomático, se concedió el alta. En enero de 2009 es visto nuevamente en forma ambulatoria por episodio febril de mayor duración de lo habitual.
DISCUSIÓN Y COMENTARIOS
Se trata de un paciente con fiebre de origen desconocido, de evolución periódica, en quien la búsqueda etiológica orientada a elementos infecciosos, inmunes y tumorales resultó negativa.
El primer elemento diagnóstico es la presentación clínica, que es la clásica de FMF, con episodios febriles de inicio en la infancia, recurrentes, autolimitados, que duran pocos días, con compromiso de serosas.
Se han descrito dos fenotipos; el fenotipo I, es el más frecuente, se caracteriza por fiebre y serositis y en ocasiones amiloidosis (el que corresponde a nuestro paciente); y el fenotipo II, en el cual la amiloidosis es la primera o única manifestación de la enfermedad (21).
El segundo elemento diagnóstico fundamental es el origen poblacional del paciente. Se trata de un descendiente de armenios tanto por línea materna como paterna. Determinadas mutaciones en el gen MEVF tienen una elevada frecuencia en ciertas poblaciones de la cuenca mediterránea, particularmente los armenios (22).
Los antecedentes familiares son otro dato diagnóstico a tener en cuenta ya que la sintomatología de su hermano se debe interpretar como causada por la misma afección. El patrón de presentación es característico de la herencia autosómica recesiva.
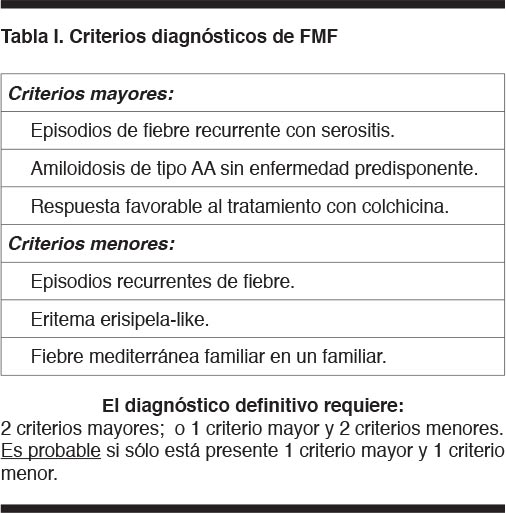
El diagnóstico de fiebre mediterránea familiar se establece en base a los criterios de Tel Hashomer (ver Tabla 1). Salvo la amiloidosis, que está ausente en la evaluación realizada, el paciente presenta todos los criterios diagnósticos.
En relación con el diagnóstico diferencial se deben plantear otras fiebres periódicas hereditarias (23). A pesar de algunas similitudes entre éstas, hay marcadas diferencias clínicas, genéticas y fisiopatológicas. En este caso las características de los episodios clínicos, la evolución y el origen poblacional del paciente son fuertemente indicadores de la FMF.
La gran mayoría de los casos publicados en la bibliografía internacional, así como los dos casos publicados previamente a nivel nacional (24,25), son de presentación en niños, edad en la que muchas veces existen solamente ataques febriles atípicos (22). En este paciente el diagnóstico tardó varias décadas, a pesar de la presentación clínica florida de la enfermedad. En nuestro medio, es habitual que se presenten dificultades en el diagnóstico ya que no se piensa en ella a la hora de plantear los diagnósticos etiológicos de una fiebre de origen desconocido y se sospecha que se trata de una enfermedad que está frecuentemente subdiagnosticada.
Con respecto a los casos y series de casos reportados en la literatura, nuestro paciente ha tenido una presentación clínica y evolución bastante típicas: crisis febriles acompañadas de peritonitis, artralgias y con menos frecuencia erisipela. En muchos casos el diagnóstico también demoró años en realizarse y hay reportados casos los cuales, al igual que nuestro paciente, fueron intervenidos ante el planteo de cuadro agudo de abdomen.
Buades en 1986 publicó un caso de una paciente española de 64 años con crisis febriles y dolor abdominal quien fue apendicectomizada a los 22 años, colecistectomizada a los 34 años y ooforectomizada a los 36 años, con persistencia de la sintomatología (21).
En 2012, Sukran Erten et al. reportaron, en Turquía, 3 hermanos: uno de ellos sufría fiebre y dolor abdominal y otro erisipela, ambos desde la juventud, permaneciendo largo tiempo sin diagnóstico (26).
Gersoni-Baurch et al., informaron de 13 pacientes con FMF en una familia consanguínea, todos ellos padecían peritonitis, pleuritis y fiebre, y tres de ellos tenían artritis o artralgias (27).
Kutlay et al. notificaron dos hermanas, que a pesar de tener, como se espera, el mismo genotipo, presentaban características clínicas diferentes: una de ellas tenía dolor abdominal intermitente, artritis y fiebre durante los ataques, mientras que en la otra predominaba la pleuritis y fiebre (28).
I. Koné-Paut et. al. (en Francia) obtuvieron los datos de 94 pacientes con diagnóstico clínico de FMF y portadores de al menos un alelo mutado del gen MEFV; presentaban en su mayoría fiebre, peritonitis, artralgias. Erupciones cutáneas en un 20% y dolor abdominal en 12% (29).
Ah Leum Lim y cols., describieron en 2012 el primer caso de FMF detectado en Korea, se trataba de un hombre de 32 años de edad, con historia de fiebre recurrente y dolor abdominal de 3 años de evolución. En una de las internaciones se realizó TC de abdomen que mostró engrosamiento edematoso de la pared del yeyuno proximal con escasa ascitis. A los tres días los síntomas remitieron al igual que el engrosamiento intestinal (30).
M. La Regina et al. publicaron en 2003 datos de 71 pacientes italianos con diagnóstico de FMF, la fiebre se presentó en el 92% de los pacientes, el segundo síntoma fue el dolor abdominal 91%. Un 22% presentó erisipela. El 72% respondió favorablemente a la colchicina. En un bajo porcentaje se evidenció compromiso renal. Uno de los pacientes fue sometido a 13 cirugías abdominales antes del diagnóstico (31).
Ante la sospecha clínica de FMF está indicado el estudio genético molecular, el cual confirmaría el diagnóstico de hallarse mutaciones en homocigosis o heterocigosis compuesta en el gen MEVF. Sin embargo si solo se demuestra mutación en uno de los alelos no se puede excluir la enfermedad, ya que el estudio no abarca todas las mutaciones del gen. Habitualmente se buscan las más frecuentes (M694V, V726A, V680I, E148Q, V694I), lo que representa un 70-80% de los pacientes. En las poblaciones de mayor prevalencia se han identificado portadores asintomáticos con mutaciones en los dos alelos lo que demuestra una gran variabilidad en la expresión clínica (32). El estudio genético, tiene además valor pronóstico, ya que la mutación M694V se asocia a un mayor riesgo de amiloidosis renal (33). También permite el estudio de otros miembros de la familia asintomáticos (o paucisintomáticos), la detección de heterocigotos y el diagnóstico prenatal. En este caso el estudio genético todavía no se ha realizado.
En cuanto al tratamiento se basa en: el control de las crisis, en evitar su aparición y disminuir el riesgo de desarrollar amiloidosis. Para el tratamiento sintomático de las crisis se emplean los antiinflamatorios sistémicos. El fármaco de primera línea es la colchicina, propuesta por primera vez por Goldfinger en 1972 (34), que inhibe la quimiotaxis de los neutrófilos, ha demostrado mejorar el pronóstico de la enfermedad, consiguiendo una remisión completa de las crisis en un 60% de los pacientes y una disminución de la frecuencia en el 30% (35). En pacientes con proteinuria, el tratamiento con colchicina a dosis más altas, disminuye la excreción de proteínas y evita la progresión del daño renal. En este caso se inició el tratamiento con colchicina a dosis de 1 mg/día, durante aproximadamente un año, sin que se presentaran episodios de fiebre desde el inicio de la terapia, con mejoría del estado general y de la calidad de vida. Esta respuesta terapéutica es un último criterio diagnóstico fundamental.
Un 5-10% de los pacientes no responden a la terapia convencional, en ellos las terapias propuestas son: el interferón alfa y la adición de colchicina intravenosa al tratamiento oral. Se ha reportado también el uso de talidomida (36), infliximab (37), inhibidores de la recaptación de serotonina (38), etanercept (39) y anakinra (40).
BIBLIOGRAFÍA
1. Janeway TC, Mosenthal HO. An unusual paroxysmal syndrome, probably allied to recurrent vomiting, with a study of the nitrogen metabolism. Trans Ass Am Phys 1908; 23: 504-18.
2. Heller H, Kariv J, Sherf L, Sohar E. Familial Mediterranean fever. Harefuah 1955; 48(5): 91-4.
3. Vergara C. Síndromes Autoinflamatorios. Rev. chil. reumatol. 2008; 24(4):206-211.
4. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. [en linea].Cell 1999[acceso 20/05/2013];97(1):133-44. Disponible en: http://ac.els-cdn.com/S0092867400807217/1-s2.0-S0092867400807217-main.pdf?_tid=faa53018-c1b7-11e2-9016-00000aab0f27&acdnat=1369100850_6ab0ae2045fc630126652a813d42b348
5. Simon A, van der Meer J, Drenth J, Firestein. Familial Autoinflammatory Síndromes. En: Kelley’s Textbook of Rheumatology, 8th ed. Philadelphia : WB Saunders Company; 2008. Chapter 113.
6. Inocencio J.Síndromes febriles recurrentes en pediatría. [en línea]. En: 34º Congreso Argentino de Pediatría, Córdoba; octubre de 2006. [acceso: 20/05/2013] Disponible en: http://www3.sap.org.ar/staticfiles/actividades/congresos/congre2006/conarpe34/material/reu_inocencio.pdf
7. Armenian Population in the World.. [en línea]. Glendale: Armenia Diaspora; s.d. [acceso 15/10/2013]Disponible en: http://www.armeniadiaspora.com/population.html.
8. Drenth JPH, van der Meer JWM, Hereditary periodic fever. N Engl J Med. 2001; 345: 1748-57.
9. Bakkaloglu A. Familial Mediterranean fever. Pediatr Nephrol. 2003; 18:853–859.
10. Orbach H, Ben-Chetrit E. Familial Mediterranean fever. A review and update, Minerva Med 2001; 92: 421-30.
11. Meiorin S, Espada G, Rosé C. Actualización. Enfermedades febriles periódicas en pediatría. Arch. Argent. Pediatr . 2006; 104:30-38.
12. Dodé C, Pécheux C, Cazeneuve C, Cattan D, Dervichian M, Goossens M, et al. Mutations in the MEFV gene in a large series of patients with a clinical diagnosis of familial Mediterranean fever. Am J Med Genet 2000; 92: 241-6.
13. Babior BM, Matzner Y. The familial Mediterranean fever gene--cloned at last. N Engl J Med. 1997;337(21):1548-9.
14. Infevers Database (The registry of MEFV sequence variants). [Base de datos en línea]. 2001c;[acceso julio 2009]. Disponible en: http://fmf.igh.cnrs.fr/ISSAID/infevers/disease_menu.php?n=1
15. Guz G, Kanbay M, Ozturk MA. Current perspectives on familial Mediterranean fever. Curr Opin Infect Dis. 2009;22(3):309-15.
16. The International FMF Consortium, Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997; 90(4):797-807.
17. French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997; 17(1):25-31.
18. Chae Jae Jin, Wood Geryl, Masters Seth L., Richard Katharina, Park Grace, Smith Brian J et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1 production. Proc Natl Acad Sci USA. 2006; 103(26): 9982-87.
19. Simon A, van der Meer JW. Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am J Physiol Regul Integr Comp Physiol 2006; 292:R86-R98.
20. van der Hilst JC, Simon A, Drenth JP. Hereditary periodic fever and reactive amyloidosis. Clin Exp Med. 2005;5(3):87-98.
21. Buades Reines, J. Fiebre mediterránea familiar. Presentación de un caso. Revisión literatura española[en línea]. Med. Balear. 1986; [acceso: 15/10/2013] 5: 25-7.Disponible en: http://ibdigital.uib.es/greenstone/collect/medicinaBalear/import/1986_v01_n2/Medicina_Balear_1986v1n2p025.pdf.
22. Yepiskoposyan L, Harutyunyan A. Population genetics of familial Mediterranean fever: a review. Eur J Hum Genet 2007; 15(9):911-6.
23. Farasat S, Aksentijevich I, Toro JR. Autoinflammatory diseases: clinical and genetic advances. Arch Dermatol 2008; 144(3):392-402.
24. Canzani R, Fischer TM, Álvarez Martínez J. Fiebre Mediterránea Familiar. Día Méd Urug 1965; XXXII (388): 597-605.
25. Mañé Garzón F, Raggio V. Fiebre mediterránea familiar: una afección frecuentemente subdiagnosticada. Rev Med Urug 2006; 22: 231-235.
26. Sukran Erten, Cahide Erzurum and Alpaslan Altunoglu. Three Family Members with Familial Mediterranean Fever Carrying the M694V Mutation Showed Different Clinical Presentations. Intern Med. 2012; 51: 1765-68.
27. Gershoni-Baruch R, Shinawi M, Shamaly H, Katsinetz L, Brik R. Familial Mediterranean fever: the segregation of four different mutations in 13 individuals from one inbred family: genotypephenotype correlation and intrafamilial variability. Am J Med Genet. 2002; 109: 198-201.
28. Kutlay S, Sengul S, Keven K, Erturk S, Erbay B. Two sisters with FMF: lack of corralation between genotype and phenotype. J Nephrol. 2006; 19: 104-10729 Kone´ -Paut I Hentgen V, Guillaume-Czitrom S, Compeyrot-Lacassagne S, Tran, TA, Touitou I. The clinical spectrum of 94 patients carrying a single mutated MEFV allele. Rheumatology (Oxford) 2009; 48:840–842. 30. Lim AL, Jang HJ, Han JW, Song YK, Song WJ, Woo HJ, et al. Familial Mediterranean Fever: The First Adult Case in Korea. J Korean Med Sci. 2012; 27(11): 1424–1427.
31. La Regina M, Nucera G, Diaco M, Procopio A, Gasbarrini G, Notarnicola C. Familial Mediterranean fever is no longer a rare disease in Italy. Eur J Hum Genet. 2003;11:50-6.
32. Shohat M, Halpern GJ, Familial Mediterranean Fever.. En: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam, M, editors. GeneReviews [En línea]. Seattle (WA): University of Washington, Seattle; 1993 [acceso: julio 2009]. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1227/
33. Ben-Chetrit E. Familial Mediterranean fever (FMF) and renal AA amyloidosis. Phenotype-genotype correlation, treatment and prognosis. J Nephrol. 2003; 16: 431-4.
34. Goldfinger SE, Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287(25):1302.
35. Kallinich T, Haffner D, Niehues T, Huss K, Lainka E, Neudorf U et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. 2007;119(2):e474-83.
36. Seyahi E, Ozdogan H, Masatlioglu S, Yazici H. Successful treatment of familial Mediterranean fever attacks with thalidomide in a colchicine resistant patient. Clin Exp Rheumatol. 2002; 20(4, Suppl 26):S43-4.
37. Metyas S, Arkfeld DG, Forrester DM, Ehresmann GR, Infliximab Treatment of Familial Mediterranean Fever and Its Effect on Secondary AA Amyloidosis. J Clin Rheumatol. 2004; 10(3):134-137.
38. Onat AM, Ozturk MA, Ozcazar L, Ureten K, Kaymak SU, Kiraz S, et al. Selective serotonin reuptake inhibitors reduce the attack frequency in Familial Mediterranean Fever. Tohoku J Exp Med 2007; 211: 9-14.
39. Mor A, Pillinger MH, Kishimoto M, Abeles AM, Livneh A. Familial Mediterranean fever successfully treated with etanercept. J Clin Rheumatol. 2007; 13(1):38-40.
40. Roldan R, Ruiz AM, Miranda MD, Collantes E. Anakinra: new therapeutic approach in children with Familial Mediterranean Fever resistant to colchicines. Joint Bone Spine. 2008; 75(4):504-5.

