










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Medicina Interna
Print version ISSN 0250-3816On-line version ISSN 1688-423X
Arch. Med Int vol.35 no.2 Montevideo July 2013
Caso clínico de interés
Síndrome de Kearns-Sayre, presentación de un caso clínico
Kearns-Sayre Syndrome
Dra. Luciana Nario
Residente de Medicina Interna, Clínica Médica 2, Facultad de Medicina. UdelaR. Montevideo
Dra María Inés Gutiérrez
Residente de Medicina Interna, Clínica Médica 2, Facultad de Medicina. UdelaR. Montevideo
Dra. Carolina Pérez
Residente de Medicina Interna, Clínica Médica 2, Facultad de Medicina. UdelaR. Montevideo
Resumen
El síndrome de Kearns Sayre es una mitocondriopatía caracterizada por disfunciones multiorgánicas que clásicamente se desarrolla antes de los veinte años de edad. Esta rara enfermedad fue descrita en 1958 por Thomas P. Kearns y George P. Sayre a través del reporte de un caso que presentaba la tríada clínica de oftalmoplejia externa, retinopatía pigmentaria y bloqueos de la conducción cardíaca; siendo esta última alteración la que determina el pronóstico. Aún no se cuenta con un tratamiento curativo para esta enfermedad. En este artículo presentamos el caso clínico de una paciente de 39 años a la que se realizó el diagnóstico de Kearns-Sayre.
Palabras clave: Síndrome de Kearns Sayre, Mitocondriopatía, Oftalmoplejia externa, Ragged red.
Abstract
The Kearns Sayre syndrome is the result of abnormalities in the mitochondria characterized by multiorgan dysfunctions, which develops before the age of twenty. This rare illness was described by Thomas P. Kearns y George P. Sayre in 1958 through the report of a case which presented the clinical triad, external ophthalmoplegia, pigmentary retinopathy and cardiac conduction defects; being this last alteration the one that determines the prognosis. There is still no treatment for this illness. In this article, is presented a case of a 39-years-old patient to whom the diagnosis of Kearns-Sayre has been made.
Keywords: Kearns-Sayre Syndrome, Mitochondrial disease, Ophthalmoplegia, Ragged red
Introducción
Thomas P. Kearns y George P. Sayre describieron en 1958 esta rara enfermedad caracterizada por la tríada clínica clásica de oftalmoplejia externa, retinopatía pigmentaria y bloqueos de la conducción cardíaca(1).
Este síndrome pertenece al amplio grupo de las enfermedades mitocondriales y sus manifestaciones aparecen clásicamente antes de los 20 años de edad. No se encontraron en la literatura revisada registros sobre la prevalencia de esta enfermedad en la población mundial; hay algún estudio sobre la baja prevalencia de las mitocondriopatías siendo alrededor de 6 casos cada 100.000 individuos(2,3).
La etiología subyace en trastornos estructurales, bioquímicos o genéticos de la mitocondria; éstas son estructuras citoplasmáticas que, mediante el proceso de fosforilación oxidativa del metabolismo aeróbico, son responsables de la producción del ATP (adenosina trifosfato intracelular) necesario para el suministro de energía para diversas funciones metabólicas(4). El síndrome de Kearns-Sayre (SKS) es un trastorno genético causado por deleciones (pérdida de un fragmento del ADN mitocondrial), el cual es heredado por vía materna con un patrón vertical no mendeliano. La madre transmite su genoma mitocondrial a todos sus hijos, pero solamente las hijas lo heredarán a todos los miembros de la siguiente generación, y así sucesivamente. Si el número de moléculas de ADNmt dañado es relativamente bajo se produce una complementación con las moléculas de ADN normal y no se manifestará el defecto genético. Cuando el ADN afectado sobrepasa un umbral determinado se manifestará un fenotipo patogénico (efecto umbral), es decir, si la producción de ATP llega a estar por debajo de los mínimos necesarios para el funcionamiento de los tejidos, debido a la producción defectuosa de proteínas codificadas en el ADNmt, se produce la aparición de la enfermedad. El umbral se suele alcanzar cuando el porcentaje de moléculas delecionadas supera el 60%(5).
En cuanto a las manifestaciones clínicas, se verán afectados los tejidos que dependen sustancialmente del proceso de fosforilación oxidativa, con gran requerimiento energético, como el tejido muscular, el tejido nervioso, las células retinianas y las células cocleares(6,7). La tríada clínica diagnóstica característica está formada por: oftalmoplejia externa progresiva, degeneración pigmentaria de la retina y bloqueo aurículoventricular. El diagnóstico se confirma mediante estudio neuropatológico a partir de una biopsia de tejido muscular a nivel de cuádriceps, con tinción de Gomori y estudio por microscopía electrónica, en el cual se observan cambios morfológicos con las características fibras estriadas también llamadas “Ragged-red” (fibras rojas rasgadas) consideradas un marcador de daño bioquímico en la fosforilación oxidativa(8). En cuanto al tratamiento, aún no se dispone de una terapéutica curativa para esta enfermedad.
Caso clínico
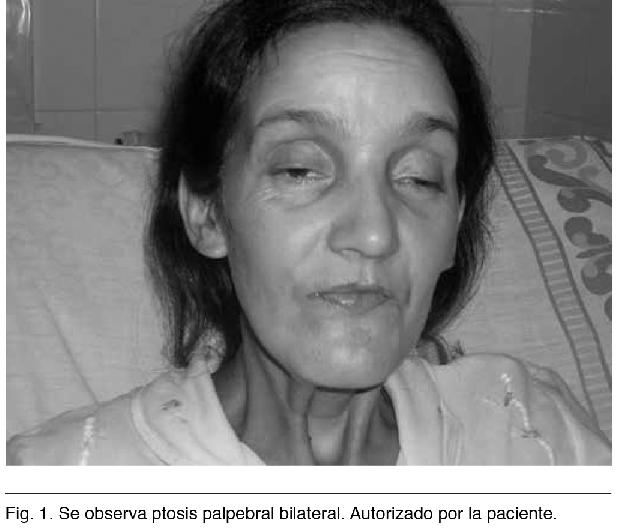
Paciente de sexo femenino, de 39 años de edad, mal medio socioeconómico. Casada, 7 hijos. Con antecedentes personales de asma intermitente desde la infancia, portadora de marcapaso definitivo desde el año 2002 (29 años de edad) por bloqueo AV completo, sin control de éste. Hipoacusia de larga data sin estudio. Ingresada al servicio de emergencia con diagnóstico de Neumonía aguda bilateral, con disfunción multiorgánica. Como antecedente de la enfermedad actual la paciente había presentado en los días previos episodio sincopal, haciéndonos pensar en el mecanismo aspirativo de la neumonía. En la internación se constata mala función del marcapaso por agotamiento de batería, procediendo a su recambio. Se implanta generador marca Biotronik modelo Talos DR tipo DDDR, se programa a modo VVI a una frecuencia de 85 ppm. La exploración física muestra, una paciente lúcida, normocoloreada, adelgazada. Al examen cardiovascular se constata ritmo regular de 80 cpm, silencios libres, normotensa. Al examen pleuropulmonar se presenta polipneica, sin cianosis; a la auscultación, estertores crepitantes bilaterales a predominio derecho, y del examen psiconeuromuscular se destaca la ausencia de movimientos oculares, ptosis palpebral bilateral (Figura 1), e hipoacusia. Fondo de ojo: despigmentación de la retina en sal y pimienta. En el sector espinal: hipotonía bilateral mayor a derecha. Leve paresia generalizada en MMSS y MMII, reflejo idiomuscular conservado, ROT conservados y reflejo cutáneo plantar en flexión bilateralmente. Sensibilidad conservada en los 4 miembros. De la paraclínica: ECG: ritmo de marcapaso, 70 cpm, QRS 0,12 seg. Ecocardiograma: diámetro de VI: 44 mm, de VD: 22 mm, FEVI 51%. Movimiento asincrónico septal. Insuficiencia tricuspídea leve. Se realizó biopsia quirúrgica de músculo estriado a nivel de cuádriceps derecho, para estudio neuropatológico bajo tinción con hematoxilina-eosina y tricrómico de Gomori, informada como: tejido muscular proximal con alteraciones histopatológicas compatibles con miopatía mitocondrial. El estudio de microscopía electrónica en el servicio de anatomía patológica, informa: fibras musculares atróficas con disrupción miofibrilar focal, megamitocondrias y fibras alteradas con acúmulos de glucógeno (Ragged Red) (Figura 2).

Dados los hallazgos clínicos y paraclínicos se realizó diagnóstico de Síndrome de Kearns-Sayre, presentando buena evolución desde el punto de vista infeccioso, asintomática desde el punto de vista cardiovascular, con buen funcionamiento de marcapaso, por lo cual fue dada de alta, coordinada para seguimiento ambulatorio en policlínica de medicina interna.
Discusión y Comentarios
Nos encontramos ante una paciente que presenta los síntomas clásicos de esta enfermedad, con inicio antes de los 20 años, como está descrito en la literatura; presentando como primera manifestación la oftalmoplejia externa progresiva, agregando hipoacusia y alteraciones en la conducción cardíaca en los años siguientes, requiriendo la colocación del marcapaso definitivo. En el síndrome de Kearns-Sayre se afectan los tejidos con mayor requerimiento energético, como el tejido muscular, el tejido nervioso, las células retinianas y las células cocleares, dando lugar a los síntomas característicos de las enfermedades que alteran el ADN mitocondrial(6,7). Se puede asociar también a retraso mental, ataxia, sordera, debilidad muscular y trastornos endócrinos, como diabetes mellitus o hipotiroidismo. La oftalmoplejia progresiva crónica externa es un grupo de hallazgos clínicos caracterizados por una inmovilidad ocular bilateral lentamente progresiva y usualmente simétrica, por lo que los pacientes tienden a no quejarse de diplopía. La afección cardíaca en el SKS es frecuente, pero con una presentación clínica variable y con diversos niveles de gravedad. Estas anomalías están presentes en 57% de los pacientes con este síndrome. Entre las principales manifestaciones cardíacas se encuentra el síncope, en 45% de los casos, la muerte súbita, en 23% y la miocardiopatía, en 20%. Estas alteraciones son las que determinan el pronóstico y la mortalidad de estos pacientes(9,10).
Se destaca el retraso diagnóstico en nuestra paciente, no siendo interpretadas las manifestaciones clínicas como parte de un síndrome. Creemos que esto se debe a la escasa familiarización de los médicos con las formas de presentación clínica de las mitocondriopatías. En este caso contamos con la posibilidad de confirmar el diagnóstico clínico planteado mediante la realización de estudio histopatológico de tejido muscular estriado, obtenido por biopsia de cuádriceps. No pudiendo realizar estudio genético por PCR de ADN mitocondrial, dado sus alto costo. Queremos enfatizar la afectación del tejido de conducción cardíaca, como manifestación frecuente y grave, ya que la mortalidad, como se mencionó previamente, está vinculada a estas alteraciones.
En cuanto al tratamiento de esta enfermedad se han realizado algunos ensayos clínicos con vitaminas, tales como vitamina C, vitamina E, tiamina, así como también con creatina, L-carnitina y coenzima Q10; lamentablemente hasta la actualidad ninguno de estos tratamientos ha mostrado buenos resultados(11). La conducta médica debe estar dirigida a mejorar la calidad de vida de los pacientes mejorando su estado nutricional, corrigiendo quirúrgicamente la ptosis palpebral, fisioterapia de rehabilitación y, si el caso lo requiere, implantar marcapaso cardíaco. En cuanto a esto, según la American Heart Asociation, se recomienda el implante de marcapaso definitivo con indicación de clase IC en pacientes con bloqueo AV 3° y 2° aun asintomáticos y clase IIb ante cualquier grado de bloqueo(12).
En nuestro caso clínico se dirigió el tratamiento a mejorar la calidad de vida, promoviendo la adecuada alimentación y el control periódico con el servicio de cardiología, con el fin de detectar tempranamente disfunciones del marcapaso, teniendo en cuenta que provenía de un medio socioeconómico y cultural deficitario, y adaptándolo a sus posibilidades. Además, se tramitó un apoyo económico, recibiendo actualmente una pensión por discapacidad.
Conclusiones
Son las mitocondriopatías enfermedades que presentan síndromes clínicos característicos, pero dada su baja frecuencia en la población, son poco conocidas entre los médicos. Siendo ésta la razón de los diagnósticos tardíos, realizándose cuando se presentan alteraciones graves, como lo son las manifestaciones cardíacas. Por esto creemos de gran importancia la difusión de estos casos clínicos y reconocerlos frente a las primeras manifestaciones, con el fin de actuar tempranamente, mejorando el pronóstico vital y funcional de estos pacientes.
Agradecimientos
A la Dra. Rosana Robaina, Dra. Daniela Fernández, Prof. Adj. Dra. Mercedes Perendones, Dra. Mesa del servicio de neuropatología del Hospital de Clínicas y Prof. Dr. Carlos Dufrechou, quienes nos apoyaron en nuestro trabajo.
Bibliografía
1. Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmoplegia and complete heart block: unusual syndrome with histologic study in one of two cases. Arch Ophthalmol. 1958;60:280-9.
2. Chinnery PF, Johnson MA, Wardell TM, SinghKler R, Hayes C, Brown DT et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol 2000; 48:188-193.
3. Arpa J, Cruz-Martínez A, Campos Y, et al. Prevalence and progression of mitochondrial diseases: a study of 50 patients. Muscle Nerve 2003; 28:690.
4. Johns DR. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N Engl J Med. 1995;333(10):638-44. Comment
5. Solano, Abelardo; Playán, Ana; López-Pérez, Manuel J. y Montoya, Julio. Enfermedades genéticas del ADN mitocondrial humano. Salud pública Méx (online) .2001,vol.43,n.2,pp.151.161. http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0036-36342001000200010&lng=es&nrm=iso>. ISSN 0036-3634
6. Ramírez-Miranda A, Navas-Pérez A, Gurria-Quintana L, Vargas-Ortega J, Murillo- Correa C, Zenteno JC. (PCR-based detection of heteroplasmic deleted mitochondrial DNA in Kearns-Sayre syndrome). Arch Soc Esp Oftalmol. 2008;83(3):155-9. Spanish.
7. Zago Filho L., Shiokawa N. Síndrome de Kearns-Sayre: relato de dois casos. Arq Bras Oftalmol. 2009;72(1):95-8.
8. Berenberg RA, Pellock JM, DiMauro S, Schotland DL, Bonilla E, Eastwood A, et al. Lumping or splitting? 'Ophthalmoplegia-plus' or Kearns-Sayre syndrome? Ann Neurol. 1977;1:37-54.
9. Síndrome de Kearns Sayre incompleto, M. A. Santolaria López, J. J. Aráiz Burdio, B. Villanueva Anadon I. Gutiérrez Cia y A. Millastre Benito. Servicio de Medicina Intensiva. Hospital Clínico Universitario Lozano Blesa. Zaragoza
10. Wallace DC. Diseases of the mitochondrial DNA. Ann Rev Biochem. 1992; 61:1175-1212.
11. Kornblum C, Schröder R, Müller K, Vorgerd M, Eggers J, Bogdanow M, et al. Creatine has no beneficial effect on skeletal muscle energy metabolism in patients with single mitochondrial DNA deletions: a placebo-controlled, double-blind 31P-MRS crossover study. Eur J Neurol. 2005;12(4):300-9.
12. Gregoratos G, Abrams J, Epstein AE, Freedman RA, Hayes DL, Hlatky MA, et al. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: Summary article.(ACC/AHA/NASPE Committee to Update the 1998 Pacemakers Guidelines). Circulation. 2002;106:214561.

