










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Medicina Interna
versión impresa ISSN 0250-3816versión On-line ISSN 1688-423X
Arch. Med Int vol.35 no.1 Montevideo mar. 2013
Caso clínico de interés
Cuando el motivo de consulta es debilidad muscular
Patients seeking care for muscle weakness
Dra. Sabrina Ventura
Residente de Clínica Médica. Facultad de Medicina. UdelaR. Montevideo.
Dra. Paula López
Asistente de Clínica Médica. Facultad de Medicina. UdelaR. Montevideo.
Dr. Arturo Pazos
Profesor adjunto de Clínica Médica. Facultad de Medicina. UdelaR. Montevideo.
Dr. Ernesto Cairoli
Profesor Agregado de Clínica Médica. Facultad de Medicina. UdelaR. Montevideo.
Dr. Juan Alonso
Profesor director de Clínica Médica. Facultad de Medicina. UdelaR. Montevideo.
Recibido: 10/09/12 - Aceptado: 08/03/13
Trabajo de la Clínica Médica “C”. Facultad de Medicina UdelaR. Hospital de Clínicas. Montevideo.
Correspondencia: Dr. Ernesto Cairoli, Clínica Médica “C” Hospital de Clínicas p. 8, Avd. Italia S/N 11600 Montevideo. E-mail: ernesto.cairoli@gmail.com
RESUMEN: Arch Med Interna 2013 - 35(1):15-18
Se analiza el caso clínico de una paciente de 48 años que consulta por debilidad muscular. Se enfatiza la importancia del examen neurológico para definir el planteo diagnóstico. Se esquematiza el razonamiento clínico en un paciente con síndrome miopático y los estudios paraclínicos a solicitar. A propósito de este caso clínico se discute el sreening diagnóstico para cáncer oculto en pacientes con miopatía inflamatoria tipo polimiositis y las posibilidades terapéuticas.
Palabras clave: síndrome miopático, miopatía inflamatoria, polimiositis, cáncer oculto.
ABSTRACT: Arch Med Interna 2013 - 35(1):15-18
This report analyses a clinical case of a 48 years old woman with muscular weakness. The relevance of neurology exam to define the diagnosis is emphasized. Clinical reasoning in a patient with myopatic syndrome and its studies are schematized. Screening for occult malignancy in a patient with polymiositis and its therapy is discussed.
Keywords: myopatic syndrome, inflammatory myopahty, polymyositis, occult malignancy.
Introducción
La aproximación diagnóstica a un paciente que consulta por debilidad muscular constituye un desafío para el médico. El examen neurológico es el eje del diagnóstico. En las neuropatías y enfermedades de la motoneurona periférica los reflejos osteotendinosos suelen estar abolidos, mientras que en las miopatías se encuentran conservados. Las fasciculaciones son un signo de lesión de la motoneurona periférica. Las alteraciones sensitivas indican neuropatía. El agotamiento muscular con el ejercicio es característico de las enfermedades neuromusculares. La miotonía es específica de algunas miopatías. La presencia de mialgias no es de utilidad para diferenciar entre una miopatía y una neuropatía(1). Se presenta a continuación el caso clínico de una paciente que consulta por debilidad muscular.
CASO CLÍNICO
Mujer de 48 años, sin antecedentes familiares a destacar; fumadora de 10 cigarillos/día, índice paquete-año (IPY) 19. HTA sistémica en tratamiento con dieta hiposódica con buen control de cifras tensionales. Prolapso de válvula mitral diagnosticada hace 20 años con ecocardiograma reciente que documentó ausencia de insuficiencia mitral y fracción de eyección del ventrículo izquierdo (FEVI) conservada. Menopausia hace un año. Papanicolau 2 meses previos al ingreso: normal. Consulta por disminución progresiva de fuerzas de miembros inferiores (MMII) a predominio proximal, de 4 meses de evolución, agregando disminución progresiva de fuerzas de miembros superiores (MMSS) a predominio de cintura escapular. No disfagia, disnea, ptosis palpebral ni otras alteraciones oculomotoras. Mialgias. No fasciculaciones. Niega consumo de fármacos o drogas. No lesiones cutáneas ni fotosensibilidad. Adelgazamiento de 5 kg en los últimos 3 meses. Niega anorexia astenia y adinamia. Tránsito urinario y digestivo sin alteraciones.
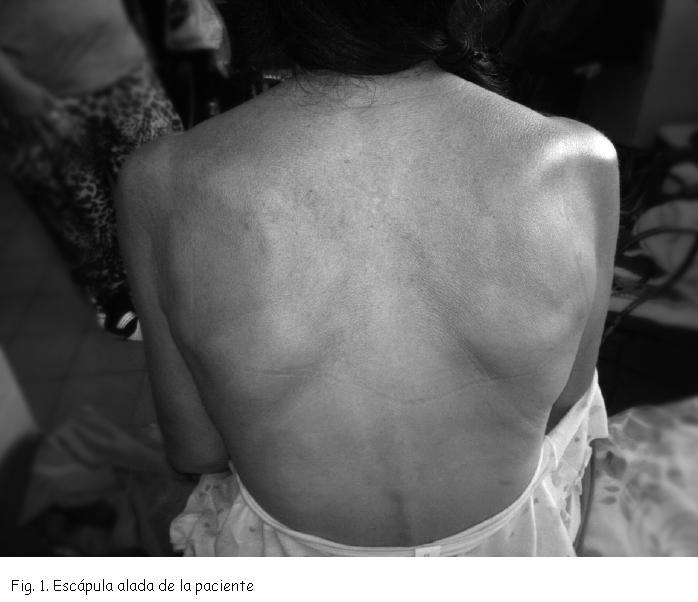
En el examen físico destaca: adelgazada, apirética, eupneica. Sin lesiones cutáneas en áreas de fotoexposición. Livedo reticularis. Sin adenomegalias. Cuello: Sin bocio. Mama: Sin alteraciones. Examen neurológico: vigilia, funciones de alta integración, pares craneanos y sector meníngeo sin alteraciones. Sector espinal: MMSS y MMII: atrofia muscular proximal. Escápula alada (Figura 1). Sin fasciculaciones espontáneas o provocadas. Hipotonía. Paraparesia proximal que vence gravedad y no opone resistencia. Reflejo idiomuscular disminuido. ROT conservados. Sensibilidad y coordinación normal. Cutáneo plantar en flexión bilateral. No apokamnosis. Marcha en báscula. Resto del examen físico: sin alteraciones.
Ante una paciente con debilidad muscular proximal y progresiva de los 4 miembros, se debe plantear si su origen es miopático o neurogénico. La presencia de atrofia muscular, ausencia de reflejo idiomuscular y ROT conservados orientan a la presencia de un síndrome miopático.
De la paraclínica se destaca: creatín quinasa total (CPK): 6288U/L, glutámico piruvato transaminasa (GTP): 125 U/L, glutámico oxalacético transaminasa (GOT): 186 U/L. Electromiograma (EMG): compromiso miogénico proximal de los cuatro miembros, con mayor preservación de los reclutamientos musculares distales. Intensa actividad espontánea, fibrilación y ondas positivas, en músculos proximales que permite plantear proceso agudo.
La elevación de enzimas de lesión muscular y las alteraciones presentes en el EMG confirman el diagnóstico de miopatía. La ausencia de antecedentes familiares, la edad de la paciente y el patrón clínico de presentación orientan a una miopatía adquirida. Dentro de las miopatías adquiridas, ante la ausencia de evidencia clínica de alteraciones endócrino-metabólicas y de ingesta de tóxicos y fármacos, se plantea una miopatía inflamatoria. En vistas a confirmar el carácter inflamatorio de dicho proceso se realiza una biopsia muscular que describe la presencia de necrosis e intenso infiltrado linfocitario endomisial con las características de tejido muscular proximal con alteraciones histopatológicas correspondientes a miopatía inflamatoria del tipo de polimiositis.
Del análisis clínico y paraclínico se concluye que la paciente presenta una miopatía inflamatoria adquirida idiopática de tipo polimiositis (PM), sin evidencias en la biopsia muscular de la existencia de una dermatomiositis (DM) o una miopatía por cuerpos de inclusión (MCI), las otras dos entidades que deben ser consideradas en el diagnóstico diferencial en esta paciente. Ante el diagnóstico de PM se solicita paraclínica para valorar enfermedad neoplásica o autoinmune subyacente: velocidad de eritrosedimentación (VES): 72 mm/hora, marcadores tumorales: CEA, CA 19-9, CA15-3, CA125, B2 microglobulina: normales. Tomografía de tórax, abdomen y pelvis sin alteraciones y mamografía normal. En la valoración inmunológica se detectaron anticuerpos antinucleares (ANA) positivos 1/640, patrón moteado, siendo negativos los anticuerpos contra antígenos nucleares extraíbles, destacando especialmente la negatividad para anti-JO-1.
Se inicia tratamiento con corticoides vía oral, utilizando prednisona 1 mg/kg/día, obteniendo mejoría clínica subjetiva y objetiva progresiva, logrando incorporarse de la cama y deambular sin ayuda a los 15 días de tratamiento. Normalización de las enzimas de lesión muscular. Se administraron suplementos de calcio y vitamina D vía oral en vistas al mantenimiento de una corticoterapia prolongada. Se otorga alta hospitalaria con seguimiento en policlínica de Medicina Interna.
DISCUSIÓN y comentarios
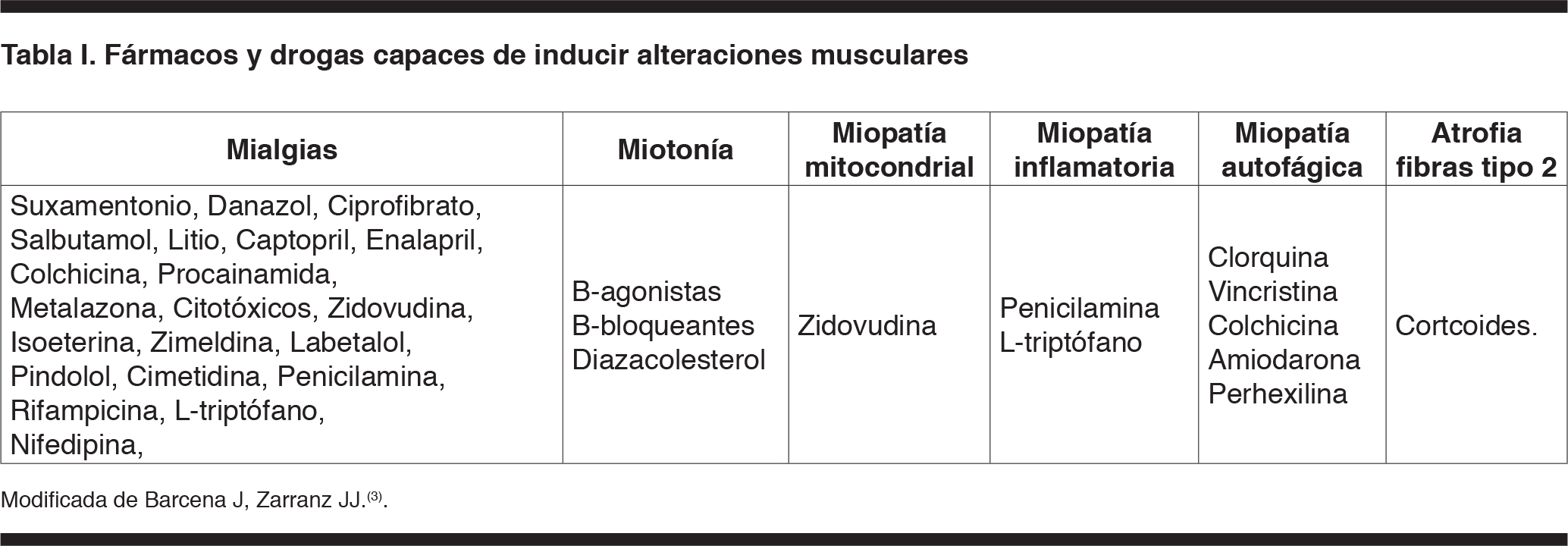
Las miopatías son un grupo heterogéneo de enfermedades en las que se afecta primariamente el tejido muscular esquelético caracterizadas por debilidad muscular y amiotrofia, pudiendo ser hereditarias o adquiridas(2,3). Las miopatías hereditarias se manifiestan generalmente en la infancia y adolescencia; se caracterizan por presentar antecedentes familiares de la enfermedad. Desde el punto de vista clínico se clasifican en distróficas y no distróficas, de cinturas o distales, con o sin afectación facial, ocular o faríngea y con o sin fenómeno miotónico. Su presentación en la edad adulta obliga a plantear el diagnóstico diferencial con miopatías adquiridas. En el caso clínico analizado se plantea una miopatía adquirida por la edad de presentación, la ausencia de antecedentes familiares y las características del cuadro clínico. Las miopatías adquiridas pueden estar etiológicamente relacionadas con patologías endocrinológicas como el hiper e hipotiroidismo, hiper o hipo paratiroidismo, síndrome de Cu-shing o enfermedad de Addison. Pueden estar asociadas a patología metabólica-tóxica como la desnutrición severa, insuficiencia renal crónica, hipopotasemia, hipermagnesemia, déficit de vitamina E, consumo de alcohol, tóxicos o fármacos (Tabla I) o finalmente encontrar en su etiología un proceso inflamatorio(2.3). Las miopatías inflamatorias se subdividen en aquellas de causa conocida y desconocida o idiopáticas. Las de causa conocida pueden ser secundarias a un proceso infeccioso de etiología viral, principalmente por el virus del VIH-1, bacteriana, parasitaria o fúngica, o estar asociadas al consumo de fármacos: zidovudina, d penicilamina, ácido 13 cis-reitinoico, estatinas. La miopatía por estatinas tiene una incidencia de 0,1 a 1,0%, es dosis dependiente y su incidencia aumenta cuando están prescriptas con otros agentes miotóxicos(4)
Dentro de las miopatías inflamatorias idiopáticas se encuentran la PM, DM y la MCI(3). Aunque la ausencia de manifestaciones cutáneas como el eritema en heliotropo y el signo de Gottron alejan el planteo clínico de la existencia de una DM, el diagnóstico definitivo es histopatológico, dado que esta entidad puede diagnosticarse aún en ausencia de manifestaciones cutáneas al obtener una biopsia compatible con DM. En cuanto a la MCI, la misma no se manifiesta con la magnitud clínica y enzimática que presentó la paciente analizada; esta entidad debe ser considerada en aquellos pacientes mayores de 50 años, más frecuentemente hombres, con debilidad muscular progresiva a predominio distal en MMSS y proximal en MMII, bajo movimiento enzimático, descripción de pocas alteraciones inflamatorias en la anatomía patológica y mala respuesta al tratamiento inmunodepresor(5).
Frente a la sospecha clínica de una miopatía inflamatoria, la solicitud de enzimas musculares se impone, siendo la CPK una de las más sensibles, en especial para la PM. Los niveles de CPK son menores o inclusive en ocasiones normales en DM asociadas a enfermedades autoinmunes sistémicas o en los casos de MCI. No obstante la CPK se encontrará elevada en los casos de PM en sufrimiento, siendo un buen marcador de actividad de la misma. Acompañando la elevación de la CPK, pueden encontrarse incremento de los niveles plasmáticos de enzimas como la TGO, TGP, LDH y aldolasa(5). Los anticuerpos específicos de miopatía inflamatoria, anti Jo-1, anti SRP y anti Mi-2 se detectan hasta en el 50% de los pacientes(6-8). Además de utilidad diagnóstica tienen valor pronóstico dado que sus niveles se correlacionan con la severidad de la enfermedad(7).
La realización de una resonancia magnética muscular, si bien no se practica de forma rutinaria, sería de utilidad para identificar los grupos musculares mayormente afectados y seleccionar el sitio más apropiado para realizar una biopsia muscular(5).
La biopsia muscular es fundamental para establecer el diagnóstico de miopatía inflamatoria observándose áreas de necrosis, signos de regeneración, aumento del tejido conectivo endo y perimisial e infiltrado inflamatorio(2). La realización de la biopsia muscular es un paso clave en la toma de decisiones, ya que la misma tiene implicancias diagnósticas, terapéuticas y pronósticas. A nivel diagnóstico puede concluir si se trata de una DM, PM o MCI. En la PM es característica, pero no patognomónica, la infiltración endomisial por células inflamatorias, predominando los linfocitos T CD8, macrófagos y el fenómeno de invasión parcial(6.8). En caso de estar frente a una DM, la búsqueda de una neoplasia subyacente es la regla debido a su mayor asociación con cáncer a diferencia de los pacientes con PM(9). Frente al hallazgo de una MCI, es necesario conocer de antemano la pobre respuesta a los inmunodepresores, pudiendo evitar la insistencia terapéutica y los efectos adversos de estas terapias.
La PM puede ser primaria o secundaria a enfermedad neoplásica o inmunológica(2.3). Hill et al.(10) realizaron un estudio descriptivo, retrospectivo y multicéntrico en 914 pacientes con diagnóstico de PM y 618 con DM según los criterios de Peter y Bohan; la frecuencia de cáncer en pacientes con PM fue de 15%. Los cánceres más frecuentemente asociados fueron linfoma no Hodgkin y cáncer de pulmón y vejiga. El mayor riesgo de presentación de la enfermedad oncológica se encuentra durante el primer año del diagnóstico de PM, siendo igual al de la población general a los 5 años(9-11). Este estudio no tiene en cuenta la histopatología por lo que la frecuencia de PM puede estar sobrestimada. El intervalo con mayor probabilidad de reconocimiento del tumor se encuentra entre los 2 años previos y los 3 años posteriores al diagnóstico de miositis(9). El riesgo disminuye sustancialmente cada año. En pacientes con PM no se ha observado incremento del riesgo de carcinoma escamoso y adenocarcinoma(9). Son factores de riesgo para desarrollo de cáncer: edad avanzada, sexo masculino, miositis refractaria o recurrente y debilidad muscular severa incluyendo compromiso de músculos respiratorios y disfagia(9). La presencia de enfermedad pulmonar intersticial y anti Jo-1 se asocian a menor desarrollo de neoplasias (9). La enfermedad tumoral agrava el pronóstico de la miositis. No existe acuerdo en los estudios paraclínicos a solicitar en un paciente con PM sin evidencia clínica de enfermedad neoplásica. El screening rutinario en búsqueda de cáncer no se asocia a detección de enfermedad maligna en etapa temprana. La comparación de las pruebas de screening rutinarias versus un screening exhaustivo ha demostrado que la TC tóraco-abdominal en el hombre, y tóraco-abdómino-pélvica en la mujer, es el estudio de mayor utilidad en estos pacientes(9). El incremento del CA 125 y CA 19-9 se asocia con mayor riesgo de cáncer(9). Algunos autores proponen solicitar analítica general con perfil básico de sangre y bioquímica, marcadores tumorales de próstata, ovario, mama, TC toráco-abdómino-pélvica, mamografía y exploración ginecológica(6). La utilización de PET/TC con fluordeoxiglucosa versus el screening convencional: examen físico completo, hemograma y bioquímica completa, TC tóracoabdominal, CEA, CA125, CA19-9; PSA, examen ginecológico, ecografía ginecológica y mamografía, tiene igual sensibilidad y especificidad para el diagnóstico de cáncer oculto en pacientes con PM y DM(12). Por lo tanto no se demuestra beneficio adicional con la realización de PET/TC (12).
Los glucocorticoides son la primera línea terapéutica. Mejoran la fuerza muscular pero no modifican la sobrevida (3,6,13). Se inician a dosis altas, prednisona 1 mg/kg/día, durante las primeras 6 semanas para establecer el control de la enfermedad, con reducción progresiva posterior hasta la dosis efectiva más baja. El tratamiento total es de 9 a 12 meses. La azatioprina, metotrexato y micofenolato de mofetilo se utilizan como tratamiento de segunda línea asociados a los corticoides(3.613). L.a evidencia para apoyar o refutar el uso de gammaglobulina intravenosa en el tratamiento de la PM es insuficiente(14). El ejercicio físico moderado junto al tratamiento inmunosupresor mejora la performance muscular al reducir la expresión de factores proinflamatorios y profibróticos(6). Las nuevas perspectivas terapéuticas están dirigidas hacia los anti-TNF y anti-INF-alfa(13).
BIBLIOGRAFÍA
1. Sarnat H. Enfermedades neuromusculares. En Berhman RE, Kliegman RM, Jenson HB. Nelson tratado de pediatría. 16a ed. Madrid: Mc Graw Hill; 2000. p. 2032-34.
2. Cervera C. Miopatías (enfermedades musculares). En Codina Puiggros A. Tratado de Neurología. 10ª ed. Madrid: Libro del Año; 1994. p. 867-909.
3. Barcena J, Zarranz JJ. Enfermedades musculares y de la unión neuromuscular. En Zarranz JJ. Neurología. 4ta ed. Madrid: Elsevier; 2008. p. 707-39.
4. David W, Chad D, Kambadakone A, Hedley-Whyte ET. Case records of the Massachusetts General Hospital. Case 7-2012. A 79-years-old Man with Pain and weakness in the Legs. N Engl J Med. 2012; 366: 944-54.
5. Dalakas, M.C. Therapeutic advances and future prospects in immune-mediated inflammatory myopathies. Ther Adv in Neurol Disord. 2008 Nov;1(3): 157-66.
6. Selva-O´Callaghan A, Trallero E. Miopatías inflamatorias. Dermatomiositis, Polimiositis, Miositis por cuerpos de inclusión. Reumatol Clin. 2008; 4(5): 197-206.
7. Miopatías inflamatorias. En Victor M, Adams RD, Ropper AH. Principios de Neurología de Adams y Victor. 7ma ed. México: Mc Graw Hill; 2002. p 1387-1937.
8. Rayavarapu S, Coley W, Nagaraju K. An update on pathogenic mechanisms of inflammatory myopathies. Curr Opin Rheumatol. 2011; 23: 579-84.
9. Aggarwal R, Oddis ChV. Paraneoplastic myalgias and myositis. Rheum Dis Clin N Am. 2011; 37: 607- 21.
10.Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 2001; 357: 96-100
11.Kuo CF, See LC, Yu KH, Chou IJ, Chang HC, Chiou MJ. Incidence, cancer risk and mortality of dermatomyositis and polimyositis in Taiwan: a nationwaide population study. Br J Dermatol. 2011:165(6): 1273-79.
12 Selva-O´Callaghan A, Grau JM, Gámez C, Vidaller A, Martínez X, Trallero E, et al. Conventional cancer screening versus PET-CT in dermatomyositis / polymyositis. Am J Med. 2010; 123(6): 558-62.
13.Hakl AE, de Paepe B, de Bleecker JL, Tak PP, de Visser M. Dermatomyositis and polymyositis: new treatment targets on the horizon. Neth J Med 2011; 69(10): 410-21.
14. Patwa HS, Chaudhry V, Katzberg H, Rae-Grant AD, So YT. Evidence-based guideline: Intravenous immunoglobulin in the treatment of neuromuscular disorders. Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2012; 78: 1009-1015.


{kind=link}
{kind=link}