










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Medicina Interna
Print version ISSN 0250-3816On-line version ISSN 1688-423X
Arch. Med Int vol.34 no.2 Montevideo 2012
Caso clínico de interés
Oftalmoplejia dolorosa, un desafío diagnóstico:
a propósito de un caso clínico de Tolosa-Hunt
Painful ophthalmoplegia, a diagnostic challenge:
on a case of Tolosa-Hunt Syndrome
Dr. Mario Torales
Profesor Adjunto Clínica Médica 2. Facultad de Medicina. UdelaR. Montevideo.
Dra. María Noel Olivera
Residente Medicina Interna Hospital Pasteur. Facultad de Medicina. UdelaR. Montevideo.
Dra. Amelia Olazarri
Residente Medicina Interna Hospital Pasteur. Facultad de Medicina. UdelaR. Montevideo.
Dr. Carlos Dufrechou
Profesor Director de Clínica Médica 2. Facultad de Medicina. UdelaR. Montevideo.
Resumen: Arch Med Interna 2012 - 34(2): 60-63
El clínico frente a una oftalmoplejia dolorosa, debe encarar diferentes alternativas diagnósticas, donde la disciplina semiológica y los estudios imagenológicos, son de capital importancia para arribar al diagnóstico. El síndrome de Tolosa-Hunt es una probable etiología, donde el uso de critérios diagnósticos, el estudio por resonancia magnética y la remisión con tratamiento corticoideo, son fundamentales para llegar al mismo, sabiendo que el seguimento evolutivo es de suma importancia para descartar los diagnósticos diferenciales.
Palabras clave: Oftalmoplejia dolorosa, Síndrome de Tolosa-Hunt.
Abstract: Arch Med Interna 2012 - 34(2): 60-63
The clinician faced with a painful ophthalmoplegia, must deal with diagnostic alternatives where semiotic discipline and imaging studies are of paramount importance to arrive at diagnosis. The Tolosa-Hunt syndrome is a likely etiology, where the use of diagnostic criteria, magnetic resonance study, and remission with corticosteroid therapy are essential to achieve diagnosis, knowing that the evolutionary track is very important to rule out various differential diagnoses.
Keywords: Painful ophthalmoplegia, Tolosa-Hunt syndrome.
INTRODUCCIÓN
La oftalmopatía dolorosa se caracteriza por presentar dolor periorbitario y hemicraneo del mismo lado, se agrega compromiso ipsilateral de III par, con afectación de la rama sensitiva oftálmica del V par. En esta entidad existen una amplia gama de entidades, difíciles de diferenciar, ya que comparten atributos clínicos. Una de las etiologías probables, es el Síndrome de Tolosa-Hunt (STH)(1).
El STH es una rara afección , siendo su incidencia de 1 caso por millón anual, se caracteriza por ser una oftalmoplejia dolorosa; puede ser recurrente; generalmente unilateral, con parálisis oculomotora ipsilateral, de etiología idiopática debida a una inflamación granulomatosa no específica del seno cavernoso o del ápex de la órbita, con rápida respuesta al tratamiento corticoideo (1). Esta afección determina una amplia gama de planteos diferenciales, uno de los más difíciles, es con el pseudotumor orbitario (PTO), dado que también se afectan los pares craneanos, tiene carácter recidivante, responde a los corticoides; la diferencia radica en la localización estando este último, confinado a la órbita, siendo el diagnóstico solo imagenológico (1,2).
Algunos autores describen, que puede ser la misma entidad, debido a que se ha documentado pacientes con ambas afecciones; además de no hallarse evidencia clínica- terapéutica para tal distinción(1,2).
La oftalmoplejia dolorosa constituye un desafío diagnóstico para el clínico que obliga a descartar enfermedades de etiología muy dispar, ya sea traumática, vascular, infecciosa, autoinmune, neoplásica, las que pueden presentarse de forma similar.
Es el objetivo de este trabajo, presentar un caso de oftalmoplejia dolorosa, analizar las dificultades diagnosticas y realizar una revisión del tema, haciendo énfasis en el STH.
CASO CLÍNICO
Paciente de sexo femenino, 36 años, con antecedentes personales de síndrome depresivo, tratada con ansiolíticos y sertralina. Consulta por un cuadro de 20 días de evolución de instalación espontanea y progresiva de dolor retro y periorbitario intenso derecho, que calma parcialmente con analgésicos no esteroideos, acompañado de cefalea no pulsátil, gravativa de hemicraneo derecho.
Exámen físico: Vigil, orientada con funciones superiores indemnes; pares craneanos: III par - asimetría palpebral dada por ptosis derecha, estrabismo divergente a derecha, parálisis del músculo recto interno y superior, sin compromiso de la oculomotricidad intrínseca; V par - hipoestesia del territorio de la rama oftálmica del trigémino derecho. Fondo de ojo: normal. Resto de examen clínico no presenta elementos destacables.
Los exámenes de laboratorio solicitados fueron: recuento hematológico, glicemia, azoemia, creatininemia, uricemia, ionograma, hepatograma, test de función tiroidea, todos, sin elementos particulares a destacar. También se solicitó pruebas para descartar enfermedades reumatológicas y del colágeno que fueron negativas. Test de sífilis, serología para el virus de inmunodeficiencia adquirida (VIH). El estudio del líquido cefalorraquídeo mostró presión de apertura, proteínas, glucorraquia,y celularidad dentro de límites normales. La tomografía de tórax y electrocardiograma no presentaron hallazgos patológicos .
Con planteo de oftalmoplejia dolorosa de posible etiología vascular o tumoral se solicita:
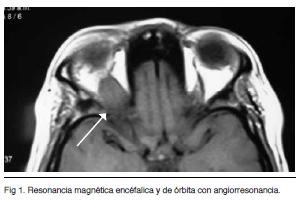
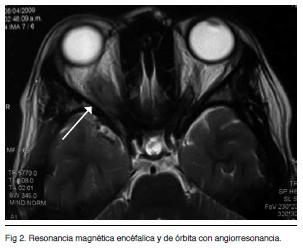
Resonancia magnética encéfalica y de órbita con angiorresonancia (Figuras 1 y 2)
Identifica un proceso sólido que ocupa el vértice de la órbita derecha. Compromete el musculo recto superior y desplaza discretamente la vena orbitaria superior. Se presenta isointenso en las secuencia T1 y T2 con realce luego de la administración del contraste i/v. La secuencia vascular, luego de la del contraste, no observó alteraciones vasculares destacables. Probable Pseudotumor orbitario o probable STH derecho.
Tratamiento y evolución
Dada la clínica, mas los hallazgos imagenológicos de STH, se inicia tratamiento con prednisona vía oral a dosis de 60 mg día con desaparición del dolor a las 48 hs del inicio de la terapia, y de la oftalmoplegia a la semana posterior; actualmente se realiza el seguimiento ambulatorio en el servicio externo de neurología.
DISCUSIÓN
El Síndrome de Tolosa Hunt es una enfermedad de rara ocurrencia, descrita en 1954 por el español neurocirujano Eduardo Tolosa y en 1961 Steven Hunt de Ohio, describe el síndrome en forma más completa, determinando los elementos clínicos más sobresalientes de esta afección.
Es más frecuente en el sexo femenino (relación mujer/ hombre 2/1), se presenta en la edad media de la vida, como en el caso expuesto(3).
El STH constituye solo el 2,9% a 3,4% de las oftalmoplejias dolorosas (OD), y solo es bilateral en el 4,1% de los casos(4). Clínicamente se caracteriza por dolor orbitario, este puede preceder a la oftalmoplejia o aparecer días después, generalmente es progresivo y precede en 2 semanas la neuropatía, se describe como un dolor retroocular “aburrido” constante, como “roer”, puede ser más intenso lancinante; se acompaña de diplopía, y en el examen neurológico se puede hallar el compromiso del III (85%), VI (70%), IV (29%) y la rama oftálmica del V, siendo muy típicos de la afección. En la paciente se describe el compromiso del III par y el V (rama oftálmica) par craneano(5,6).
Las fibras simpáticas periarteriales se pueden comprometer y producir un Síndrome de Horner que solo ocurre en el 20% de los casos, al igual que el nervio óptico pero no es lo común(6).
Se considera que es un proceso inflamatorio inespecífico, de tipo granulomatoso, que compromete el seno cavernoso, fisura orbitaria, ápex de la órbita, si bien se han descrito otras localizaciones inusuales como la temporo- parietal o hemicraneo(5,6).
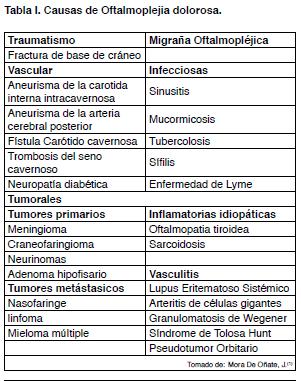
Otros diagnósticos diferenciales de OD, que responden a los corticoides: son el meningioma, tumor de células gigantes, fístulas carótidea-cavernosa, adenoma pituitario, Granulomatosis de Wegener, sarcoidosis, y migraña oftalmoplejica(6).
Los aneurismas del sector cavernoso rara vez causan dolor, se excluyen por arteriografía cerebral, los tumores de la región se diferencian por RMN (resonancia magnética), infiltran y se produce edema de las estructuras del seno cavernoso. La sarcoidosis, linfoma y meningioma aparecen hipointensa o isointensa en el etapa temprana en tomografía de contraste(5,6).
Tanto la sarcoidosis como el linfoma se acompañan la mayoría de las veces, de síntomas sistémicos.
En la Granulomatosis de Wegener, oftalmoplejia del diabético, migrañosa y los aneurismas no se halla masa intracavernosa, ni en el ápex de la órbita, a diferencia de lo que ocurre en el STH(6).
El dolor que ocurre en la trombosis séptica del seno cavernoso es súbito, y se acompaña de síntomas infecciosos sistémicos, en el estudio por RMN se observa un engrosamiento del seno cavernoso, con un área hipointensa que puede corresponder al trombo sanguíneo(6).
Dada la amplia gama de afecciones descritas en el diagnostico diferencial (Tabla I) se formularon una serie de criterios diagnósticos, entre ellos los de la sociedad internacional de cefalea en 1988, y los cuales fueron modificados en 2004, y son los que determinan el diagnostico de STH(7,8). Estos criterios son:
- Uno o más episodios de dolor o orbitario unilateral persistente durante semanas de no mediar tratamiento.
- Parálisis de uno o mas de los nervios craneales III, IV y/o VI y/o demostración de un granuloma por la RNM o biopsia
- La parálisis coincide con el inicio del dolor, o le sigue en un intervalo de dos semanas
- El dolor y la paresia desaparecen en 72 hs. cuando se trata adecuadamente con esteroides
- Se han excluido otras causas
Se han reportado casos en donde la rama oftálmica del trigémino está afectada en su primera división como en la paciente que se analiza; el compromiso del nervio facial, el óptico y el acústico, también han sido descrito. En algunos casos también se observó la afectación de la inervación simpática de la pupila, pero no es frecuente. En el caso de la paciente, que se trae a consideración, se cumplen la mayoría de los requisitos para realizar diagnostico de STH(6).
La tomografía de cráneo de alta resolución puede mostrar cambio en la densidad de los tejidos profundos del seno cavernoso y de la fisura superior de la órbita, pero es menos sensible que la RMN, dado los artefactos producidos por el tejido óseo(6,10).
Al momento actual la RMN es el examen de primera línea para diagnostico y seguimiento; siendo posible visualizar un engrosamiento asimétrico del seno cavernoso pudiendo ser isointenso en las imágenes ponderadas en T1 y T2 respecto a la sustancia gris, como en el estudio de la paciente. Existe impregnación de la lesión con el uso de medio de contraste paramagnético. Además se puede observar una disminución del calibre de la porción de la carótida interna intracavernosa comprometida, como también una convexidad anormal en la pared del seno cavernoso(10).
El estudio imagenológico mencionado, puede orientar a enfermedades de la órbita ocular, muchas veces con presentación clínica similar, como la oftalmopatia tiroidea, o síndromes linfoproliferativos. En el caso del PTO los vientres musculares y los tendones de la órbita se engrosan en forma uniforme adquiriendo aspecto de túbulo, en cambio en la oftalmopatia tiroidea los tendones están indemnes. Puede ser más difícil el diagnóstico cuando el engrosamiento es de un solo musculo orbitario, donde el mayor diagnostico diferencial es la infiltración tumoral(11).
El estudio del LCR permite descartar otras entidades que cursan con síndrome del seno cavernoso como meningitis infecciosas y neoplásicas.
La analítica en el LCR para descartar las diferentes probabilidades diagnósticas, incluye la serología para sífilis, enfermedad de Lyme, enzima convertidora de angiotensina, hongos y micobacterias. Si bien puede existir leve pleocitosis e hiperproteinorraquia, estos hallazgos obligan a descartar otras patologías como neoplasma, meningitis infecciosa o inflamatoria(6).
En la imagenología pueden presentar contrastes de intensidad similares: meningiomas, sarcoidosis, diseminación local de carcinomas, y linfomas; sin embargo los meningiomas no se resuelven con corticoides, la sarcoidosis, metástasis y linfomas, en general tienen otros síntomas sistémicos(6,11).
La exploración quirúrgica del seno cavernoso con biopsia, es un método invasivo, que en este contexto reviste dificultades técnicas y no está exenta de riesgo, se plantea cuando se requiere de altas dosis de corticoides para mitigar los síntomas, cuando progresan los síntomas neurológicos(6,11).
La evolución del STH, puede remitir en forma espontanea en un 40%, ser recurrente con apariciones frecuente en meses y a veces en años(6).
El tratamiento de elección son los glucocorticoides que claramente aceleran la resolución del cuadro, si bien no existe evidencia clara de cuál es la dosis, ruta de administración, o tiempo de tratamiento más eficaz(5,10).
Si bien se recomiendan los corticoides por vía intravenosa, la vía oral puede ser una alternativa igualmente eficaz. Se inician altas dosis, por 4 a 6 semanas, siendo su retiro gradual por varias semanas o meses. Una pauta recomendada es prednisona vía oral, 80 a 100 mg el primer día, luego 60 mg al día y cada dos semanas descender, a 40, 20 y 10 mg hasta completar 2 semanas más. Si bien esta pauta resulta efectiva, se puede variar de acuerdo a la respuesta clínica del paciente(5,6).
El seguimiento ambulatorio es de extrema importancia; la monitorización de la respuesta es clínica e imagenológica a través de la solicitud de la RMN, donde la respuesta es más lenta desde el punto de vista radiológico. La RMN debe realizarse cada 2 meses hasta lograr la resolución del proceso inflamatorio, posteriormente el seguimiento deber continuar hasta los 2 años por si reaparecen síntomas o imagen patológica que obliguen nuevamente a evaluar las diferentes alternativas diagnosticas. Los corticoides resuelven el dolor en 72 hs.; el compromiso de los pares craneanos es más lento, se produce en 2 a 8 semanas y es inusual que permanezca déficit neurológico. En la evolución ¼ de los pacientes puede presentar una recaída, a los meses o años del tratamiento, en esta situación, es necesario descartar diferentes desordenes inflamatorios o neoplásicos, como Sarcoidosis, Granulomatosis de Wegener o linfoma(5,6).
CONCLUSIONES
El cuadro clínico de oftalmoplejia dolorosa constituye un desafío clínico, donde el análisis semiológico adquiere jerarquía para elaborar los diferentes diagnósticos diferenciales; los estudios imagenológicos como la RMN, permiten arribar con más precisión y a la vez, evaluar la evolución durante el tratamiento. El diagnóstico de STH se realiza a través de criterios establecidos; debiendo igualmente evaluar las diferentes enfermedades alternativas, donde la evolución y seguimiento posterior al tratamiento son de suma importancia.
Los corticoides son la terapéutica de elección; donde remisión rápida del cuadro clínico constituye un pilar diagnostico.
El pronóstico generalmente es bueno, si bien pueden observarse secuelas, en el caso descrito, la paciente presentó una respuesta funcional excelente.
BIBLIOGRAFÍA
1. Mora De Oñate J, Pascual Pérez Alfaro R, Izquierdo Vázquez C, González Ruiz M, Aguirrebeña Olmos A, Díez-Villalba R. Painful ophthalmoplegia (pseudotumor of the orbit and Tolosa-Hunt syndrome). Arch Soc Esp Oftalmol. 2007; 82: 509-512.
2. Hamad-Cueto O, Tamayo-Toledo JA, Mármol-Prados AA, García-Trujillo L, López-Madrona JC, Fernández-Fernández O. Síndrome de Tolosa-Hunt y pseudotumor orbitari: entidades solapadas en un caso con perfil clínico no habitual. Rev neurol. 2006; 42: 530-534.
3. Pascual J, Cereza L, Canga A, Alvarez de Arcaya A, Polo JH, Berciano J Tolosa-Hunt syndrome: focus on MRI diagnosis. Cephalalgia.1999; 19 Suppl25: 36-38.
4. Alioglu Z, Akbas A, Sari A, Erdöl H, Ozmenöglu Z. Tolosa Hunt síndrome : caso report. J neuroradiol. 1999; 26:68-72.
5. Kline LB, Hoyt WF. The Tolosa-Hunt syndrome . J Neurol Neurosurg Psychiatry. 2001;71:577-582.
6. Shindler K. Tolosa-Hunt syndrome. UptoDate [ en línea] versión 19.3. 2012.[ acceso: julio 2012] Disponible: http://www.uptodate.com /contents/tolosa-hunt-syndrome.
7. Colnaghi S, Versino M, Marchioni E, Pichiecchio A, Bastianello S, Cosi V, et al. ICHD-II diagnostic criteria for Tolosa–Hunt syndrome in idiopathic inflammatory syndromes of the orbit and/or the cavernous sinus. Cephalalgia 2008, 28: 577–584.
8. Mendez JA, Arias CR, Sanchez D, Pesci LM, Lopez BL, Lopez R, et al. Painful ophthalmoplegia of the left eye in a 19-year-old female, with an emphasis in Tolosa-Hunt syndrome: a case report. Cases J. 2009.17;2: 8271.
9. Monzillo PH, Saab VM, Protti GG, Costa AR, Sanvito WL. Síndrome de Tolosa-Hunt. Análise de seis casos. Arq Neuropsiquiatr 2005; 63:648-651.
10. Cakirer, S. MRI findings in Tolosa-Hunt syndrome before and after systemic corticosteroid therapy. Eur J Radiol 2003; 45(2):83.
11. de Arcaya AA, Cerezal L, Canga A, Polo JM, Berciano J, Pascual J. Neuroimaging Diagnosis of Tolosa-Hunt Syndrome: MRI Contribution. Headache 1999; 39:321-325.

