










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkArchivos de Medicina Interna
Print version ISSN 0250-3816On-line version ISSN 1688-423X
Arch. Med Int vol.33 no.3 Montevideo Dec. 2011
Casuística de interés
Púrpura de Schönlein-Henoch, presentación en el adulto
Schönlein-Henoch purpura, presentation in the adult
Dr. Rodrigo Andrade
Residente de Medicina Interna. Clínica Médica A. Facultad de Medicina UdelaR. Montevideo.Dra. Ximena Añón
Residente de Medicina Interna. Clínica Médica A. Facultad de Medicina UdelaR. Montevideo.Dra. Verónica Pérez
Especialista en Medicina Interna. Asistente de Clínica Médica. Facultad de Medicina UdelaR. Montevideo.Dr. Mauricio Amaral
Especialista en Medicina Interna. Asistente de Clínica Médica. Facultad de Medicina UdelaR. Montevideo.Dra. Laura Llambí
Especialista en Medicina Interna. Profesora Adjunta de Clínica Médica. Facultad de Medicina UdelaR. Montevideo.Dra. Cecilia Baccino
Dra. Ximena Cabrera
Dr. Alfredo Álvarez Rocha
Profesor Director de la Clínica Médica A Facultad de Medicina UdelaR. Montevideo.
Palbras clave: Vasculitis; Púrpura de Schönlein-Henoch.
Systemic vasculitis are a group of low prevalence disorders that affect vessels of ifferent caliber, having an heterogeneous clinical presentation. Among these, Schönlein-Henoch Purpura (HSP), defined as a systemic vasculitis of small vessel that present in over 90% of cases in children, with an incidence of 13,5 per 100,000 inhabitants, with a generally benign prognosis. The presentation in adulthood is rare, reaching an incidence of 0.03-0.35/100.000 per inhabitants. It typically presents with palpable purpura, arthritis, abdominal pain and renal involvement. According to different series, the presentation in children and adults is similar, having a less favorable prognosis in adults conditioned by kidney disease. Since no controlled clinical trials are available, current evidence is based solely on expert opinion. Therapeutic approach includes nonsteroidal antiinflammatory drugs (NSAID) corticosteroids, immunosuppressive drugs and in some cases plasmapheresis.
Keywords: Vasculitis. Henoch-Schönlein Púrpura
INTRODUCCIÓN
El término púrpura o síndrome de Schönlein-Henoch fue introducido por Schönlein en 1837, para describir el caso de un niño, el cual clínicamente se presento con púrpura y artritis.
Fue en el año 1874, en que Henoch añadió a esta descripción original la afectación intestinal y renal, por lo que desde ahí, el nombre síndrome de Schönlein-Henoch parece el más adecuado históricamente(1,2). Forma parte de un grupo de enfermedades donde el hecho patogénico central es la inflamación de la pared vascular, afectándose principalmente los vasos de pequeño calibre, de diferentes localizaciones, de ahí su presentación clínica heterogénea.
A pesar del tiempo transcurrido desde su descripción inicial, la etiopatogenia no es bien conocida, no disponiendose de criterios diagnósticos universalmente aceptados ni de un tratamiento médico estandarizado (1,2).
Es considerada la vasculitis más frecuente de la edad pediátrica, con una incidencia de hasta 13.5 /100.000 habitantes(3-5). Desde el punto de vista clínico, se caracteriza por la tríada de púrpura no trombocitopénico, afectación articular (artritis o artralgias) y dolor abdominal, a lo que se añade en una proporción variable de pacientes la existencia de afección renal (6,7).
En los adultos, su incidencia cae notoriamente, presentándose en 0,03-0,35/100.000 habitantes(8), siendo más frecuente en el sexo masculino que en el femenino(1,2). Tiene un pronóstico menos favorable que en la edad pediátrica, siendo la afección renal la manifestación más importante, ya que ésta marca el pronóstico y tiene un rol preponderante desde el punto de vista terapéutico.
El diagnóstico de esta entidad se basa por tanto en pilares clínicos, paraclínicos y anátomopatológicos, con demostración de la existencia de una vasculitis leucocitoclástica y característicamente por la demostración de depósitos vasculares de IgA en la inmunofluorescencia directa tanto en la piel como en cualquiera de las biopsias de los órganos afectados(9).
El objetivo de este artículo es describir un caso de PSH en la edad adulta y realizar una revisión de la literatura.
MATERIALES Y MÉTODOS
Se describirá un caso clínico de Púrpura de Schönlein Henoch, con compromiso cutáneo y renal de presentación en la edad adulta.
Se realizo un revisión de la literatura en las siguientes bases bibliográficas: PubMed, LILACS, Scielo, Springer entre 1978-2010.
CASO CLÍNICO
Hombre, de 52 años, procedente de Montevideo, con antecedentes personales de hipertensión arterial y accidente cerebro vascular isquémico en el año 2001 sin secuelas.
Consultó por cuadro de 10 días de evolución dado por dolor abdominal tipo cólico difuso y lesiones de piel en MM.II. (Figura 1) y orinas hipercoloreadas.
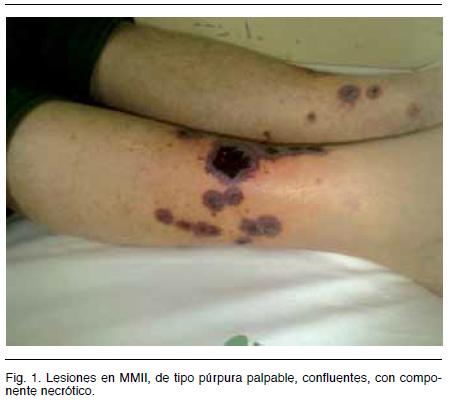
Del examen físico se destaca: lesiones de tipo púrpura palpable, confluentes, con componente necrótico y elementos inflamatorios en tercio inferior de piernas.
Resto del examen físico sin alteraciones.
De la paraclínica se destaca: hemograma sin plaquetopenia, crasis normal. Azoemia: 60 mg/dl, creatininemia: 0.97 mg/dl. Examen de orina: Hemoglobina +++, proteinuria 2.27 g/dl, cilindros hialinogranulosos.
Urocultivo negativo. Ecografía de aparato urinario normal.
Con planteo clínico de vasculitis sistémica con afección cutáneo-renal, se solicitan estudios diagnósticos complementarios, de los que se destacan: Proteinuria en 24 horas: 1,46 g/24 hs.
Dosificación de: ANA, ANCA, Ac antiMBG, crioglobulinas negativos. Complemento y PEF normales. Dosificación de IgA normal. Serología para HIV, VDRL, VHC, VHB: negativos.
FGC gastritis crónica. Sangre oculta en heces: negativa.
Biopsia de lesiones cutáneas: vasculitis leucocitoclástica.
Dada la presencia de elementos de lesión glomerular se decide realización de punción biópsica renal (Figura 2), la cual evidencio: muestra de 16 glomérulos con 12% de obsolescencia, uno de ellos evidenciaba lesión de esclerosis segmentaria, en tanto que el resto tenían un moderado aumento de la matriz mesangial así como ssas capilares obliteradas.
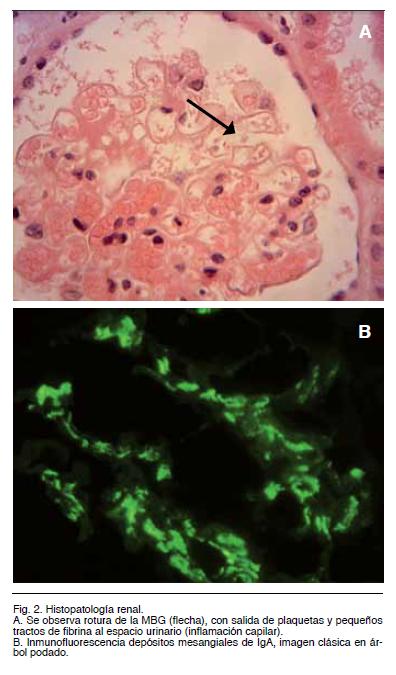
Se observa rotura de la membrana basal glomerular (Figura 2A, flecha), con salida de plaquetas y pequeños tractos de fibrina al espacio urinario. Hay escasos PMNs circulantes. El sector Túbulointersticial presenta una atrofia tubular con fibrosis intersticial de 30% de la corteza analizada. Hay moderados sectores con lesión tubular aguda, con ocasionales cilindros hemáticos y proteicos, destacándose zonas con capilares peritubulares dilatados por la presencia de abundantes glóbulos rojos. En el sector vascular hay arteriolas con hipertrofia y edema de la capa media. Otras arteriolas muestran una franca disminución de su luz, con necrosis fibrinoide parietal.
La inmunofluorescencia fue: IgA+++ mesangial, IgG difuso mesangial, IgM negativa, positiva en cilindros, C3+ positivo mesangial, fibrinógeno positivo en cailares glomerulares (Figura 2 B).
Se concluye que se trata de una Nefropatía con depósitos mesangiales dominantes de IgA, con elementos de actividad dados por roturas de asas capilares con extravasación de fibrina, esclerosis segmentaria en un glomérulo, hipercelularidad mesangial moderada, sin proliferacion endocapilar; atrofia tubular y fibrosis intersticial leve (elementos histopatológicos de cronicidad).
El dignóstico histopatológico fue concluyente para Nefropatía IgA (NIgA), el cual se hace hasta el presente exclusivamente por la demostración, en general por inmunofluorescencia, de IgA en el mesangio en forma dominante o codominante. En este caso presento un claro patrón mesagial, que es uno de los típicos de esta entidad, con una positividad de+++.
Dado la presentación clínica del paciente y los hallazgos anatomopatológicos se realiza diagnóstico de PSH.
El tratamiento se realizó en base a prednisona, 1 mg/kg/día, con descenso progresivo de los mismos, asociado a 6 bolos de ciclofosfamida mensuales, tres de 1 gramo y otros tres de 500 mg, dado el extenso compromiso renal del paciente.
La evolución fue favorable con remisión de las lesiones cutáneas, y normalización del sedimento urinario y la proteinuria.
DISCUSIÓN y COMENTARIO
El PSH de presentación en al adulto es una entidad infrecuente, nuestro caso clínico coincide con los datos de otras series, donde la edad promedio de aparición en el adulto es alrededor de los 50 años (10).
Dentro de la clásica tétrada diagnóstica, nuestro paciente presento tres: lesiones cutáneas, dolor abdominal y nefropatía, hallazgos que coinciden con las series internacionales.
El porcentaje de presentación para las lesiones cutáneas es mayor a 90%(1,9) donde, a diferencia de la edad pediátrica, pueden ser lesiones de tipo vesículas hemorrágicas o lesiones necróticas(11), como las que se describieron en este caso.
En lo que respecta a la afección abdominal, las diferentes series muestran una frecuencia de presentación en 65% de los pacientes, siendo más frecuente si existe concomitantemente compromiso renal(1,9). El dolor abdominal tipo cólico difuso es la manifestación más característica y puede acompañarse de náuseas, vómitos, diarrea, rectorragias y melenas. Estos últimos hallazgos no estuvieron presenten en nuestro paciente.
En cuanto a la afección renal, su frecuencia de presentación aumenta en la edad adulta (12), oscilando en las distintas series entre 30 y 80%(1,13). Generalmente tienen mayor riesgo de nefropatía los pacientes adultos, si se asocia además afección intestinal y lesiones cutáneas(1), como en el caso de nuestro paciente.
El espectro clínico es variado, manifestándose en 90% de los casos sólo por alteraciones del sedimento, tal como es caso clínico analizado, existiendo otras formas de presentación como oliguria, hipertensión arterial o proteinuria.
La insuficiencia renal es rara (menor a 2%) al igual que el fracaso renal extremo terminal (menor a 1%)(1).
En todos las series analizadas, la nefropatía es un marcador pronóstico del PSH, sobre todo en el adulo. Además de ser más frecuente y más grave que en la edad pediátrica, algunas series señalan la presencia de hipertensión, proteinuria mayor a 1 g/24 hs o la presencia de insuficiencia renal al inicio del cuadro como factores pronósticos en el adulto (14).
La afectación renal es dinámica y puede evolucionar con los años, incluso en ausencia de manifestaciones extrarrenales (15).
Si bien la etiología del PSH es desconocida, se cree que influyen factores ambientales y genéticos(16). Como posibles factores precipitantes, se han implicado distintos agentes infecciosos, fármacos y, ocasionalmente, alimentos y tumores.
Se ha encontrado procesos infecciosos previos, especialmente de las vías respiratorias superiores hasta en 50% de los enfermos(3,10). Además de los datos microbiológicos, el patrón estacional del PSH apoya este desencadenante infeccioso. El estreptococo es el agente infeccioso implicado más frecuentemente.
También se ha relacionado Mycoplasma pneumoniae, Legionella y Yersinia, así como al virus de la varicela, hepatitis B y C, rubeola, sarampión, citomegalovirus, parvovirus B19, adenovirus y distintas vacunas(3,4).
Los fármacos habitualmente involucrados son los antibióticos ß-lactámicos, aunque también los macrólidos y diversos analgésicos y antiinflamatorios no esteroides se han descrito como posibles agentes precipitantes (1).
La asociación con tumores parece rara; no obstante, al igual que otros síndromes vasculíticos, la PSH se ha descrito como un síndrome paraneoplásico (17,18).
Señalamos que en el caso descrito, no se encontró ningún factor precipitante.
En cuanto al diagnostico del PSH, en la última década se han propuesto hasta cuatro criterios diferentes para su clasificación(17-20).
Como puede deducirse, el problema de la definición del PSH permanece sin resolver y no hay ninguna universalmente aceptada(10). Además todas las clasificaciones utilizadas remarcan la edad (inferior a 20 años) como factor de suma importancia en el diagnóstico de PSH. Como fue expuesto anteriormente esta entidad puede presentarse en mayores de 20 años, esto llevo a la revisión de los criterios diagnósticos de l PSH, tanto que la Sociedad Europea de Reumatología Pediátrica y la Liga Europea contra el Reumatismo (EULAR/PReS), han propuesto la supresión de la edad como criterio diagnóstico, proponiendo nuevos criterios diagnósticos (Tabla I) (19).

Con respecto al tratamiento no se dispone de estudios terapéuticos controlados que permitan tomar decisiones a la hora de tratar a estos pacientes, existiendo un nivel de evidencia basada en la opinión de expertos y series de casos.
Las lesiones cutáneas habitualmente se resuelven con reposo. En casos de lesiones extensas con tendencia a la cronicidad, pueden usarse corticoides a dosis bajas o medias (15-30 mg/día de prednisona).
La artritis responde a antiinflamatorios no esteroides (AINE). Sin embargo, debido a la posible afectación digestiva o renal de estos enfermos puede ser preferible emplear corticoides a dosis bajas en lugar de AINE.
El tratamiento de las manifestaciones digestivas y renales continúa siendo controvertido.
La afectación digestiva es en general autolimitada. Glasier et al., en un estudio retrospectivo de 22 enfermos con PSH y afectación digestiva, no encuentran beneficio con el empleo de esteroides(20). En cambio, Allen et al.(21), así como Rosemblum y Winter(22), en sus estudios retrospectivos de 70 y 43 pacientes, respectivamente, encuentran que los corticoides pueden acelerar la recuperación de la sintomatología digestiva. La pauta terapéutica habitual es prednisona a dosis de 1-2 mg/kg/día por vía oral y en casos más graves metilprednisolona intravenosa. Aun en ausencia de estudios controlados, actualmente la mayoría de los autores los considera indicados si existe clínica abdominal especialmente hemorrágica(1).
El tratamiento de la nefropatía es el aspecto más debatido, existiendo en la literatura, las más variadas combinaciones de fármacos, sin un resultado con un peso estadístico significativo.
Como tratamiento inmunosupresor se han empleado corticoides orales y en bolos intravenosos, ciclofosfamida, ciclosporina, azatioprina y plasmaféresis (23).
Por tanto, hasta que se disponga de estudios prospectivos y controlados, el tratamiento del PSH, especialmente de la afectación renal, permanecerá controvertido (1).
En nuestro paciente se opto por un terapia combinada de corticoides via oral, (prednisona 1 mg/kg) con descenso progresivo, asociada con bolos de ciclfosfamida (6 en total), obteniendo una muy buena respuesta clínica, con una mejoría significativa de la función renal y del sedimento urinario (Tabla II), manteniéndose hasta la fecha asintomático, sabiendo que en esta entidad en control y seguimiento es fundamental con el fin de detectar precozmente nuevas alteraciones.
Se presentó el caso de un paciente adulto con lesiones de piel y afección renal en el cual se confirmó el diagnóstico de PSH dados los hallazgos histopatológicos en la punción biopsica renal. Se realizó tratamiento inmunosupresor en base a corticoides y ciclofosfamida con buena respuesta.
El pronóstico dependerá de la recurrencia del cuadro y de la evolución del daño renal por lo cual debe realizarse seguimiento prolongado.
Agradecimientos
Agradecemos la colaboración con fotografías y datos histopatológicos en este trabajo, a la Profesora Agregada Dra. Ana Panuncio y a la Profesora Adjunta Sylvia Melesi, pertenecientes a la Cátedra de Anatomía Patológica del Hospital de Clínicas.
BIBLIOGRAFÍA
1 Blanco, R; Rodríguez-Valverde, V; Mata-Arnaiz, C; Martínez-Taboada, V M. Síndrome de Schönlein-Henoch. Rev Esp Reumatol. 2000;27:54-65.
2 Ameal Guirado, A I; Montes Santiago, J. Unidad de Medicina Familiar y Comunitaria.Púrpura de Schönlein-Henoch en adultos: estudio de 9 casos. 1 Servicio de Medicina Interna. Hospital Meixoeiro. Vigo An. Med. Interna. (Madrid) 2004, 21(12): 79-80.
3 Saulsbury F T. Clinical Update: Henoch-Schönlein purpura. Lancet 2007;369(24):976-78.
4 Farley TA, Guillespie S, Rasoulpour M et al. Epidemiology of a cluster of Henoch-Schönlein purpura. Am J Dis Child 1989; 143: 798.
5 Steward M, Savage JM, Bell B et al. Long term renal prognosis and Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr 1988; 147: 113.
6 Cassidy JT, Petty RE. Vasculitis. En: Cassidy JT, Petty RE, editores. Textbook of pediatric rheumatology (3.a ed.). Filadelfia: W.B. Saunders, 1995; 365-422.
7 White RHR. Henoch-Schönlein Purpura. En: Churg A, Churg J, editores. Systemic vasculides. Nueva York: Igaku-Shoin, 1992; 203-17.
8 Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995; 25: 28-34.
9 Lie JT, and the Members and Consultants of the American College of Rheumatology Subcommittee on Classification of Vasculitis. Illustrated histopathologic classification criteria for selected vasculitis syndromes. Arthritis Rheum 1990; 33: 1.074-87.
10 Evange´line pillebout, Eric Thervet,Gary Hill, Corinne Alberti,Philippe Vanhille, and Dominique Nochy Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002, 13: 1271-78.
11 Wanakul S, Pongprasit P. Henoch Schönlein purpura presenting as hemorragic vesicles and bullae. Case report and literature review. Pediatric Dermatology. 1995.
12 Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García-Fuentes M, González-Gay MA. Henoch-Schönlein purpura in adulthood and childhood. Two different expressions of the same syndrome. Arthritis Reum 1997; 40: 859-64.
13 García-Porrúa C, Calviño MC, Llorca J, Couselo JM, González-Gay MA Henoch-Schönlein purpura in children and adults: clinical differences in a defined population..Divisions of Rheumatology and Pediatrics, Hospital Xeral-Calde, Lugo, Spain Semin Arthritis Rheum 2002;32(3):149-56.
14 Coppo R, Mazzucco G, Cagnoli L, Lupo A, Schena FP. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology Collaborative Study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-83.
15 Chaussain M, De Boissieu D, Kalifa G, Epelbaum S, Niaudet P, Badoual J et al. Impairment of lung diffusion capacity in Henoch-Schönlein purpura. J Pediatr 1992; 121: 12-6.
16 Frank T. Saulsbury. Henoch–Schönlein purpura. Division of Immunology and Rheumatology, Department of Pediatrics, University of Virginia Health System, Charlottesville, Virginia, USA. Current Opinion in Rheumatology 2010, 22:598–602.
17 Blanco R, González-Gay MA, Ibáñez D, López-Viana A, Ferrán C, Regueira et al. Henoch-Schönlein Purpura as clinical presentation of a myelodysplasic syndrome. Clin Rheumatol 1997; 16: 626-28.
18 Kurzrock R, Cohen P, Markowitz. Clinical manifestations of vasculitis in patients with solid tumors. Arch Intern Med 1994; 154: 334-40.
19 Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. Sociedad Europea de Reumatología Pediátrica y la Liga Europea contra el Reumatismo (EULAR/PReS) Endorsed Consensus Criteria For The Classification Of Childhood Vasculitides. Ann Rheum Dis. 2006;65:936-41.
20 Glasier CM, Siegel MJ, McAlister WH, Shackerlford GD. Henoch-Schönlein syndrome in children: gastrointestinal manifestations. Am J Roentgenol 1981; 136: 1.081-85.
21 Allen DM, Diamong LK, Howell DA. Anaphylactoid purpura in children. Am J Dis Child 1960; 99: 147-68.
22 Rosemblum ND, Winter HS. Steroid Effects on the course of abdominal pain in Henoch-Schönlein purpura. Pediatrics 1987; 79: 1.018-21
23 Hasegawa A, Kawamura T, Ito H, Hasegawa O et al. Fate of renal grafts with recurrent Henoch-Schönlein purpura nephritis in children. Transplant Proc 1989; 21: 2.130-33.


{kind=link}