











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Caso clínico
Mujer. 21 años, Sin antecedentes patológicos. Consulta por dolor abdominal gravativo de 7 meses de evolución y plenitud precoz. Examen físico normal.
En la tomografía axial computarizada (TC), se evidencia tumoración sólido quística de 70 x 58 x 55 mm, con compresión gástrica. Desplaza arteria y vena esplénica. (Fig. 1)
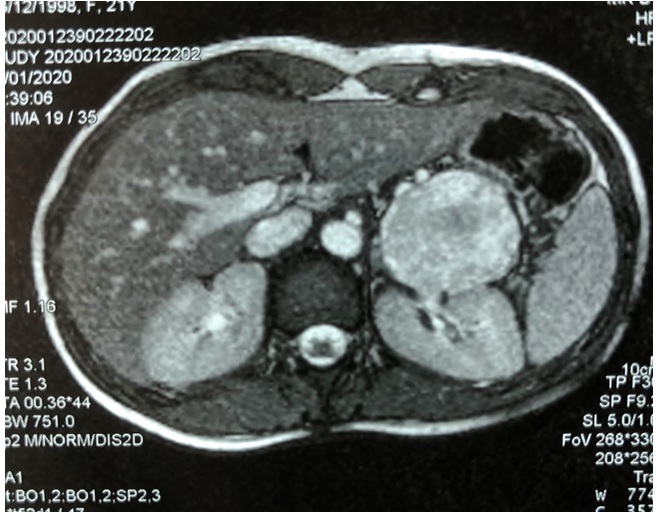
Fig. 1: Estudio TC muestra tumoración solido quística de cola de páncreas de 70x58x55mm que comprime estómago, sin infiltración. Arteria y vena esplénica permeables. Wirsung 6-8 mm. Resto del páncreas: normal.
RNM: tumor cola páncreas con patrón de proceso nodular quístico en cola de páncreas con aspecto de cistoadenoma microquístico.
Cirugía. Pancreatectomía distal y esplenectomía por laparotomía.
Buena evolución en el postoperatorio. (Fig. 2). Alta 4to.día.
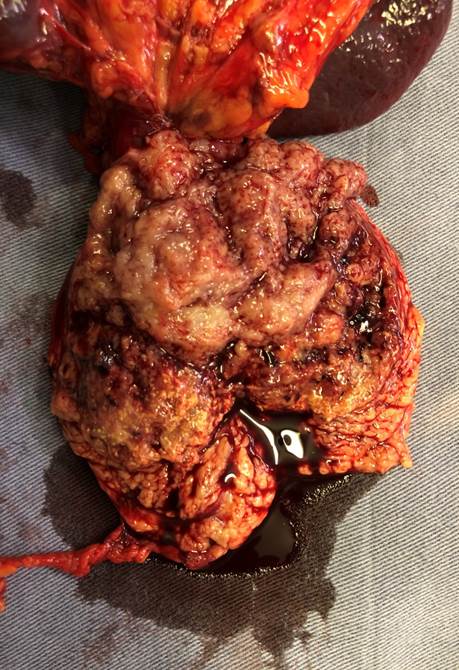
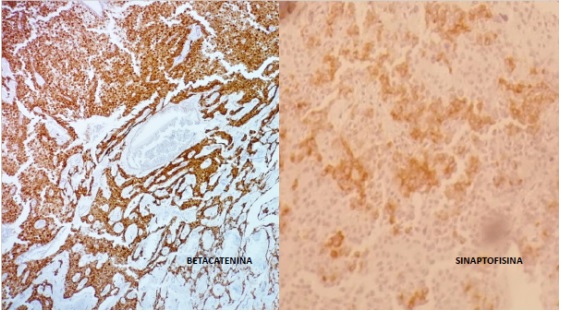
Anatomía patológica: Tumor pancreático de 56x45x35mm. Histológicamente es una neoplasia de bordes expansivos, patrón sólido, endocrinoide con sectores microtubulares, seudopapilares y estroma vascularizado. Células con núcleos regulares, con cromatina en finos grumos (en sal y pimienta) y nucléolo puntiforme poco evidente. Citoplasma escaso, finamente granular, eosinófilo o aclarado. Bajo conteo mitótico con menos de 1 mitosis en 10cga. Inmunohistoquímica (IHQ) Tinción positiva para betacatenina y sinaptofisina. (Fig. 3) con cromogranina negativa. Margen de resección y bazo, sin compromiso neoplásico .No evade la capsula del órgano. Diecinueve ganglios negativos. En suma: tumor solido seudopapilar pancreático. Seguimiento a 24 meses: asintomática.
El trabajo ha sido realizado respetando las normas internacionales sobre investigación clínica y anonimización de personas. Se cuenta con el consentimiento informado, firmado por la paciente.
La neoplasia sólida pseudopapilar es una forma rara (2 %) de tumores pancreáticos. De origen epitelial y baja malignidad; es conocido como Tumor de Franz1 o de Hammoudi2.
Fue definida como entidad clínica en 1959 por Virginia Kneeland Frantz y se consolidó como tumor pseudopapilar sólido (STP) en 1996 cuando la OMS lo incluye en clasificación de la WHO. Desde entonces y hasta la fecha se identificaron 1342 publicaciones internacionales en Pubmed - la mayoría en los últimos 20 años que reflejan un creciente interés por este excepcional tumor. Su incidencia varia ampliamente entre 0.17 % hasta 6 % de los tumores exocrinos del páncreas1)(3)(4. El 90 % de los casos afectan mujeres jóvenes en 22 y 28 años3)(4; con una relación de 6:1 sobre el sexo masculino5.
La sintomatología es tardía, por efecto de masa del tumor y crecimiento expansivo; el dolor que se constata entre el 19 % y 87 % de los casos. 6 El estudio imagenológico mediante tomografía computarizada (TC) tiene una sensibilidad del 60 % y muestra un tumor voluminoso, circunscripto con cápsula periférica bien delimitada del páncreas normal (Fig. 1) y calcificaciones en un 30 % de los casos. Estos tumores típicamente se observan como lesiones de densidad mixta, sólidas en la periferia y quísticas en el centro. Pueden existir áreas de hemorragia, como zonas de alta densidad entre la región quística y solida de la lesión. Son tumores que generalmente desplazan estructuras adyacentes más que las7)(8. La resonancia nuclear magnética (RNM) es más eficiente que la TC para detectar los cambios degenerativos como la hemorragia, cambios quísticos y la integridad de la cápsula8.
En cuanto al PETscan/F18-FDG, las lesiones con SUV alto, son altamente sospechosas de malignidad y sugieren la exéresis quirúrgica, incluso en pacientes asintomáticos. Si las células no captan la glucosa, existe alta probabilidad de que se trate de una lesión benigna que permitiría una resección más limitada9. Dependiendo del patrón imagenológico y tamaño incluso se puede considerar el control evolutivo estricto.
Se desconoce su etiología. Se ha propuesto un origen ductal epitelial, neuroendocrino, una célula primordial pluripotencial e incluso origen extra pancreático de origen genital10.
El diagnóstico definitivo es anatomopatológico con patrón solido-quístico y pseudo-papilar en proporciones variables; además se ha descrito recientemente el aumento de la expresión nuclear y citoplasmática de la E-cadherina y la beta-catenina como marcadores específicos11.
El patrón inmunohistoquímico característico muestra un patrón celular vimentina, alfa 1 antitripsina y alfa 1 antiquimiotrpsina positivas. En algunos casos expresan positividad para enolasa neuronal específica (NSE) y sinaptofisina. Con frecuencia existe la presencia de receptores de progesterona, CD10 y CD56.Marcadores de enzimas pancreáticas y marcadores endócrinos son generalmente negativos. El antígeno epitelial de membrana (EMA) y la cromogranina son negativas. Estos tumores son consistentemente negativos para cromogranina lo que ayuda a diferenciarlos de los tumores neuroendócrinos12)(9.
El papel de los marcadores tumorales en el diagnóstico y pronóstico de SPN ha sido explorado intensamente, con diversos perfiles de compatibilidad; los de mayor valor son vimentina, alfa-antitripsina, NSE y progesterona13).
Un patrón inmunohistoquímico probado es: CD56, CD10 y beta-catenina positivos fuerte. Progesterona y sinaptofisina con positividad focal, y citoqueratina AE1-AE3 y cromogranina negativos14. Otro perfil de compatibilidad es: vimentina, NSE, CD68 y alfa antiqui- mioripsina positivas y sinapto- fisina y cromoganina negativas9.
En este caso, el diagnostico se realizó en base al aspecto microscópico solido-seudopapilar y la inmunomarcación positiva para betacatenina y sinaptofisina; con negatividad para cromogranina. (Fig 3).
El diagnóstico diferencial común de SPT incluye adenoma microquístico, tumor de células de los islotes no funcionante, neoplasma mucinoso cístico, pancreatoblastoma y el pseudoquiste hemorrágico calcificado15)(16.
El 85 % de estos tumores se encuentran limitados al páncreas en el momento del diagnóstico. El resto tienen metástasis sincrónicas cuyas localizaciones más frecuentes son el hígado, ganglios regionales, el mesenterio, el epiplón y el peritoneo.
El tratamiento indicado es la resección quirúrgica incluso en presencia de metástasis viscerales. La recurrencia local/distancia se observa entre 12 a 15 %.
De la experiencia nacional encontramos cinco publicaciones, una mención en el contexto de tumores de páncreas17, un caso pediátrico, en un niño de 12 años18 y dos series breves de cuatro casos cada una9)(19.
Camblor9 en una comunicación reciente, uno de sus casos, luego de una pancreatectomía distal, tuvo una evolución con metástasis pulmonares que fueron tratadas mediante excéresis y tratamiento poliquimioterápico, con una sobrevida de 7 años. Sin embargo, no consta un claro diagnóstico de SPN histológico ni inmunohistoquímico, sino que se limita a sugerir el origen pancreático de las lesiones pulmonares. El resto de los pacientes fueron tumores voluminosos - entre 6 y 17 cm- distales en los cuales se realizó solo la excéresis quirúrgica, con excelente evolución entre 20 meses y 18 años.
Varela19 comunica también cuatro casos, de sexo femenino, con un promedio de edad de 18 años, volumen tumoral promedio de 10 cm., topografiados en cuerpo y cola del páncreas. En todos se realizó cirugía resectiva; sin complicaciones ni recidivas a 42 meses de control.
Un autor brasileño, Silano6 presenta 14 casos propios, de tumores con una media de 6.7 cm. sin metástasis. El 57 % de los casos localizados en cuerpo y cola del páncreas. Se realizó la excéresis completa en todos, acorde con la topografía del tumor. No se encontró infiltración linfática regional, pero en dos casos fue necesario realizar una interposición protésica vascular. En todos los casos la confirmación diagnóstica de SPN fue mediante inmunohistoquímica. Asimismo, realiza una revisión de trabajos retrospectivos entre 2006 y 2020, que acumulan una experiencia de 1210 casos y muestra como dato interesante que el diagnóstico incidental se realizó entre el 15 % y 65 % de los pacientes.
La gran mayoría son tumores son de muy baja agresividad, pero entre el 10 % -15 % de los casos, pueden ser francamente malignos. Los indicadores de agresividad incluyen invasión de órganos vecinos , estructuras vasculares, metástasis hepática y recidiva post tratamiento.Ciertas características histopatológicas se han asociado con un comportamiento agresivo: rotura capsular y alteración de relación solido/quística ,con mayor componente de este último , alto índice mitótico, atipias nucleares, necrosis extensa, áreas sarcomatoides y relacionadas con la expresión de Ki-6720. El Ki-67 se ha propuesto como indicador de malignidad , de forma que un índice bajo (inferior al 5 %) indica un crecimiento tumoral lento y mejor pronóstico.
Clambor9 analizando varios autores, sugiere que el rol de la quimioterapia y radioterapia no está bien definido en la literatura, existiendo pocos reportes que describen su uso. Los reportes de casos en los que SPN irresecables fueron tratados con quimioterapia y radioterapia muestran beneficio en un número limitado de pacientes. En cuanto a la quimioterapia se ha descripto el uso de Gemcitabine, 5-Fluoracilo, Adriamicina, Mitomicina C y Cisplatino.
El pronóstico es favorable aun en presencia de metástasis a distancia, con supervivencias globales de 95 % a 5 años 90 % a 10 años incluso en presencia de metástasis hepáticas o peritoneales20.
Tang20 reportó una sobrevida de 97 % en una serie de 36 pacientes resecados, incluyendo 7 pacientes con metástasis hepáticas. Igual resultado se observa en una revisión que incluyó 718 pacientes reportados en la literatura.
El interés de esta comunicación ha sido la presentación y revisión de la literatura, de un tipo excepcional de tumor sólido primitivo del páncreas, de etiología desconocida, que afecta a mujeres jóvenes, con un potencial de malignidad que puede llegar al 15 % de los casos; cuyo tratamiento es la resección completa del tumor. Su diagnóstico está basado en la inmunomarcación positiva con vimentina, betacatenina y sinaptofisiona, con cromogranina característicamente negativa. Su pronóstico es favorable, con sobrevida prolongada incluso en etapa metastásica

