











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La hiperplasia suprarrenal congénita (HSC) comprende un grupo de defectos en enzimas responsables de la biosíntesis del cortisol a partir del colesterol. Todos los tipos de HSC son monogenéticos y autosómicos recesivos1-3. Existen siete subtipos de HSC causadas por mutaciones en los genes que codifican las enzimas de la vía de la esteroidogénesis y que producirán diferentes manifestaciones clínicas: 21-hidroxilasa (21OH), 11β hidroxilasa (11β-OH), 17β hidroxilasa (17-OH), 3β- hidroxiesteroide deshidrogenasa tipo 2 (3β-HSD2), proteína reguladora aguda esteroidogénica (StAR), enzima de escisión de la cadena lateral del colesterol P450 (SCC) y P450 oxidorreductasa (POR)2-4.
La deficiencia de 21-hidroxilasa (21-OHD) es la más frecuente, representando más del 90% de los casos. De ellos, el 75% corresponde a una forma clásica perdedora de sal. A nivel internacional se reporta una incidencia de 1:14.000 a 1:18.000 nacimientos en la forma clásica, mientras que la no clásica se presenta en 1:2.000 recién nacidos5. En Uruguay la incidencia es de aproximadamente 1 cada 7.9776.
Las manifestaciones clínicas de la HSC clásica pueden deberse a una deficiencia de cortisol o aldosterona, o ambas, cuando están presentes y a la síntesis excesiva de esteroides bioactivos. En el caso de 21OHD, se producen grados variables de exceso de andrógenos (Figura 1). En pacientes con la 21OHD clásica, la exposición en el útero a un exceso profundo de andrógenos conduce a la virilización de los genitales en las niñas, que se identifica fácilmente al nacer (Figura 2). Por el contrario, los niños afectados tienen hallazgos físicos mínimos o nulos. La implementación en el recién nacido del cribado redujo de forma marcada el tiempo antes del diagnóstico4,5.
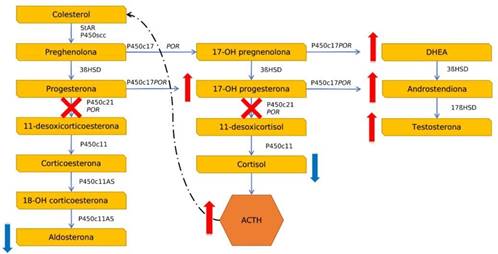
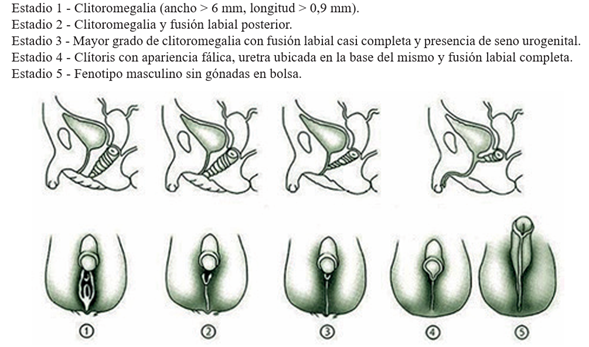
Figura 2 Estadios de Prader. Elaboración propia a partir de Rodríguez A, Sanz M, Echevarría M. Hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. Pediatr Integral 2015; XIX(7): 488-97.
En Uruguay, a partir del año 2007 se hizo obligatoria la pesquisa neonatal para la HSC7,8. El diagnóstico prenatal busca mutaciones en el gen CYP21A2 a través de la punción de vellosidades coriales o amniocentesis, también a través del ADN fetal amplificado extraído de sangre materna, que permite el diagnóstico antes de las nueve semanas y evita tratar fetos de forma innecesaria. Se recomienda en HSC con ambos padres portadores de la mutación severa y el antecedente de un hijo previo con la forma clásica, ya que se trata de una enfermedad autosómica recesiva4,9. El tratamiento prenatal se considera en etapa experimental, a través de la administración materna de dexametasona en fetos femeninos con riesgo de enfermedad clásica, manteniéndose el tratamiento hasta el parto con la confirmación diagnóstica10. El objetivo del tratamiento en el recién nacido es reducir la secreción excesiva de andrógenos, reemplazando las hormonas deficientes, y a través de la corrección de la hipovolemia; el glucocorticoide (GC) de elección es la hidrocortisona. Los mineralocorticoides (MC) (por ejemplo, fludrocortisona) pueden ayudar a descender la cantidad de GC necesaria.
La monitorización del tratamiento se realiza a través de la 17-OHP y la androstenediona en sangre2.
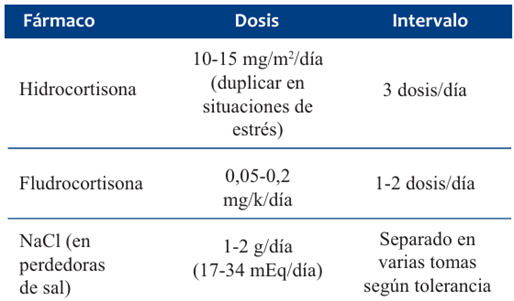
En la forma perdedora de sal tienen hiponatremia y pueden requerir aporte de sodio de 1 a 5 mEq/kg en los primeros dos años de vida. En relación con el tratamiento, no existen guías clínicas basadas en evidencia científica robusta. Según recomendaciones de expertos, la mejor pauta es realizar hidrocortisona 10 a 15 mg/m2/día en tres dosis, pues simula de forma más cercana al ritmo circadiano, si bien no está exenta de complicaciones, ya que se mantiene en dosis suprafisiológicas. También conlleva una monitorización difícil al carecer de marcadores adecuados5,11. Las complicaciones principales a largo plazo están vinculadas tanto a la enfermedad de base como a su tratamiento, y son el aumento de la mortalidad (duplica la de la población general), el riesgo de restricción de crecimiento y talla baja, la pubertad precoz o tardía, la baja densidad ósea vinculada al uso prolongado de MC, el síndrome metabólico, los trastornos del sueño y fatiga3,11.
Objetivo
Describir el caso clínico de una recién nacida con HSC clásica perdedora de sal, diagnosticada por el tamizaje de 17-OH progesterona, que concurre a consulta con elementos de shock. Se revisa la evidencia actual sobre métodos de diagnóstico y tratamiento disponible.
Caso clínico
Neonato de sexo femenino de 11 días de vida al ingreso.
De los antecedentes perinatales se destaca: madre de 26 años, sana, raza blanca. Talla 170 cm. Padre de 22 años, raza blanca. Talla 165 cm.
Producto de primera gestación. Embarazo bien controlado de captación precoz, sin complicaciones. Toxoplasmosis inmunizada. Exámenes de laboratorio del tercer trimestre: estreptococo del grupo B negativo. VDRL negativo. HIV negativo. Hepatitis B negativo. Ecografías obstétricas normales.
Parto vaginal con presentación cefálica, sin complicaciones. Se recibe una recién nacida, término maduro, adecuada, vigorosa. Peso al nacer: 3.220 g, perímetro cefálico: 35 cm, longitud: 51 cm, Apgar: 9/10.
Permanece en alojamiento conjunto madre-hijo por 48 horas, con buena adaptación a la vida extrauterina. Alta conjunta a domicilio, alimentada con pecho directo exclusivo a demanda, con buena succión y tolerancia.
A los 11 días de vida la madre concurre a primer control pediátrico, pesquisa neonatal alterada, con alteración de la 17-OH progesterona: 160,4 ng/dl (VR 0,5-5,5). Peso al momento de la consulta: 3.170 g.
Al examen físico se destaca paciente sudorosa, panículo adiposo hipoturgente, adelgazada. Piel hiperpigmentada, especialmente en área genital (Figura 3). Genitales atípicos, presentando clitoromegalia (1 cm x 1,5 cm) y fusión posterior de labios mayores (clasificación de Prader 2) (Figura 3).
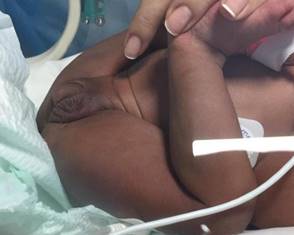
De los exámenes de laboratorio se destaca, hemograma: hemoglobina 20 g/dl, hematocrito 57%, leucocitos 14.700/ml plaquetas 606.000/mm3, glicemia 0,88 mg/dl, gasometría arterial: pH 7,42 pCO2: 25 mmHg pO2 107 mmHg BE: -8 meq/L HCO3 19 meq/L Agap 25 meq/L, lactato 4 mmol/L: 98 mEq/L K+: 7 mEq/L, Na+ 128 mEq/L.
Se realiza cariotipo para confirmación de sexo cromosómico: 46 XX.
Se realiza el tratamiento del shock con cargas de suero fisiológico a 10 ml/kg intravenoso. Ingresa al área de internación.
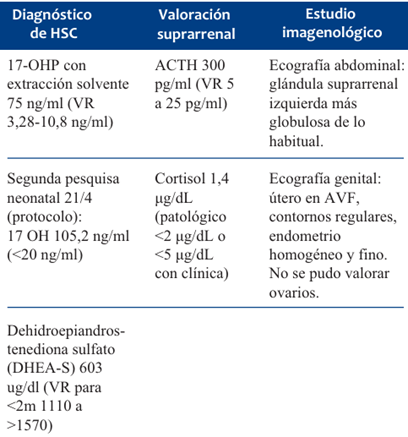
En interconsulta con endocrinología y con el planteo de HSC perdedora de sal, se solicitan análisis de laboratorio y estudios de imagen para confirmar diagnóstico y valorar repercusión (Tabla 1).
Se realizan correcciones de sodio y se inicia hidrocortisona a dosis 50 mg/m2/día.
Requiere un aporte total de sodio de hasta 2 gramos por día para lograr valores de sodio normales. A los 18 días de vida se inicia fludrocortisona 0,05 mg por día, que se aumenta a 0,1 mg por día dada la escasa mejoría en aumento ponderal y dificultad en el control de iones. Se adjuntan gráficas de evolución de iones y peso durante la internación (Figuras 4 a 6).
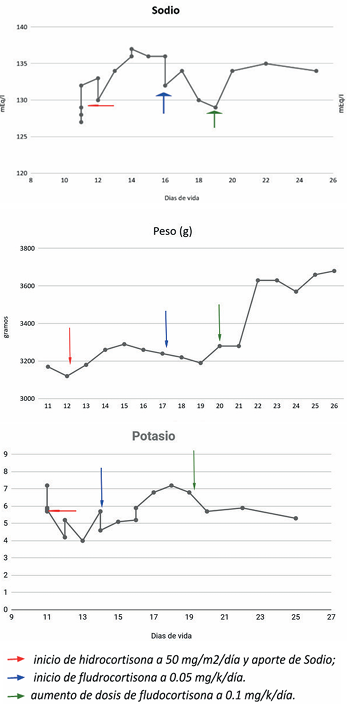
Figuras 4 - 5 - 6 Luego del Evolución del sodio, potasio y peso en el tiempo ajuste en el tratamiento específico, se logra la estabilidad en iones y un aumento ponderal mantenido, por lo que se otorga alta a domicilio con seguimiento ambulatorio.
Consentimiento para la utilización de la historia clínica
Se obtuvo el consentimiento informado de los padres para la utilización del caso clínico.
Discusión
La HSR clásica continúa siendo una enfermedad potencialmente mortal en donde la sospecha, el diagnóstico y tratamiento oportuno son fundamentales para mejorar la sobrevida de los pacientes afectados.
Si bien en niñas la clínica con virilización de genitales es característica en la HSR clásica perdedora de sal, el diagnóstico a través del examen clínico es tardío cuando se compara con el diagnóstico realizado por el tamizaje neonatal, como lo observaron Speiser PW y colaboradores en su publicación5. En el caso reportado, la paciente presenta elementos clínicos compatibles con virilización de genitales femeninos, dado por clitoromegalia y fusión labial posterior, clasificado como Prader 2; sin embargo, no se realizó el diagnóstico clínico al nacimiento. La paciente consulta por un resultado de tamizaje de la 17-OHP elevado e ingresa con alteración del medio interno, deshidratación y elementos de shock.
La alteración en genitales externos femeninos requiere obligatoriamente una fuerte sospecha clínica y un tratamiento precoz para evitar la progresión a shock y muerte de no tratarse a tiempo.
El tamizaje neonatal se ha implementado en varios países (Estados Unidos, Italia, Suecia, Austria, República Checa, Países Bajos), y en Uruguay desde 2009, con buenos resultados. La sensibilidad del screening con 17-OHP se sitúa entre 73% y 100%, y su especificidad alrededor del 100% cuando se estratifican los puntos de corte según edad gestacional o peso al nacer12. Este test puede dar falsos positivos por reactividad cruzada de conjugados esteroides, por elevaciones transitorias en el recién nacido pretérmino o bajo estrés, y tiene baja especificidad de algunos anticuerpos para 17-OHP, por lo que frente a un resultado positivo, debe confirmarse mediante la medición de 17-OHP con extracción de solvente orgánico, que elimina los factores mencionados5.
La consejería genética para los padres es recomendada con el fin de identificar mutaciones en el gen CYP21A2, y, en el caso de futuros embarazos, realizar el diagnóstico prenatal y discutir el tratamiento intrauterino9,10.
Se plantea el tratamiento prenatal con dexametasona en fetos femeninos con riesgo elevado de padecer la forma clásica. Se considera en fase experimental, existen distintos protocolos, la mayoría inicia tratamiento antes de las siete semanas en fetos con ambos padres portadores, mantiene el tratamiento en aquellos con sexo femenino hasta confirmación de portador del gen. De confirmarse, se mantiene tratamiento hasta el parto. Dado que el riesgo de padecer la enfermedad en portadores es de 1 en 8, existen dudas en cuanto al riesgo/beneficio10. El tratamiento posnatal incluye el uso de glucocorticoides y MC que suplanten el cortisol (y sus derivados) y la suplementación de sodio en las formas perdedoras de sal. En situaciones de estrés debe aumentarse la dosis de glucocorticoides, tales como enfermedades moderadas o severas, cirugías, traumas, etc. En las crisis adrenales perdedoras de sal se suma el aporte de fluidos a lo antedicho, con suero fisiológico a razón de 20 ml/k de pérdida de peso. Durante estas crisis, puede aparecer acidosis, hipoglicemia e hiperpotasemia2. Existe en el mercado internacional hidrocortisona de liberación dual (inmediata y extendida) que permite administrar una dosis diaria, con reporte de mejoría en adherencia y calidad de vida, además de simular mejor el ritmo circadiano11. La cirugía de genitales externos femeninos está discutida en la actualidad. La guía publicada en The Journal of Clinical Endocrinology and Metabolism, en 2010, recomienda considerar la cirugía de reconstrucción perineal y del clítoris en virilizaciones severas (Prader mayor o igual a 3), durante la infancia temprana11,13. En esta paciente el tratamiento inicial está dirigido a revertir el shock, corregir la acidosis y aportar gluco y MC, así como una monitorización de la acidosis, hipoglucemia, hiperpotasemia e hiponatremia.
El tratamiento de mantenimiento (Tabla 2), supervisado por especialistas en endocrinología, permitió en este caso normalizar los valores de iones y mejorar la curva de peso (Figuras 4 a 6).
Si bien no existen en la bibliografía actual nuevos fármacos para el manejo de esta patología, la optimización del tratamiento, el diagnóstico oportuno y un rápido tratamiento del shock, mineralo, glucocorticoides y aportes de sodio mejoran el pronóstico y favorecen la normalización del medio interno y el incremento ponderal, disminuyendo la mortalidad de esta grave patología endocrinológica. Persiste en discusión la oportunidad de corrección quirúrgica en el caso de genitales externos femeninos viralizados.

